, Carmen L. Menendez2, Rodolfo Montironi3 and Liang Cheng4
(1)
Department of Surgery and Pathology, University of Cordoba Faculty of Medicine, Cordoba, Spain
(2)
Pathology Department, Hospital de Cabueñes, Gijón, Spain
(3)
Department of Biomedical Sciences and Public Health, Polytechnic University of the Marche Region (Ancona), Torrette, Italy
(4)
Department of Pathology, Indiana University School of Medicine, Indianapolis, Indiana, USA
1.1 Basic Anatomy and Histology
The kidneys are paired retroperitoneal organs that normally extend from 12th thoracic vertebra to the 3rd lumbar vertebra. The average adult kidney is 11–12 cm long. It weighs 125–170 g in men and 115–155 g in women.
The three defined regions, upper polo, middle zone and lower pole usually reflect regions drained by three lobar veins.
The normal adult kidney has a minimum of 10–14 lobes, each composed of medullary pyramid surrounded by a cap of cortex.
The renal parenchyma consists of the cortex and the medulla. The cortex is the nephron- containing parenchyma. The renal medulla is divided into outer medulla and the inner medulla or papilla. The papilla protrudes into a minor calyx. Its tip has 20–70 openings of the papillary collecting ducts (Bellini ducts).
The cortex contains glomeruli, proximal and distal convoluted tubules, connecting tubules, and the initial portion of the collecting ducts, as well as interlobular vessels, arterioles, capillaries, and lymphatics. The interstitial space is scant; it contains the peritubular capillary plexus and inconspicuous numbers of interstitial fibroblasts and reticulum cells.
1.2 Overview
The current classification of renal cell tumors was proposed in 2004 by the World Health Organization (WHO) and has been recently updated by the International Society of Urological Pathologists (Table 1.1). It describes categories and entities based on pathological and genetic analyses. A number of emerging or provisional categories are also incorporated (Table 1.2)
Table 1.1
International Society of Urological Pathologists (ISUP) Vancouver modification of WHO (2004) Histologic Classification of Renal Tumors
Renal cell tumors
Papillary adenoma
Oncocytoma
Clear cell renal cell carcinoma
Multilocular cystic clear cell renal cell neoplasm of low malignant potential
Papillary renal cell carcinoma
Chromophobe renal cell carcinoma
Hybrid oncocytic chromophobe tumor
Carcinoma of the collecting ducts of Bellini
Renal medullary carcinoma
MiT family translocation renal cell carcinoma
Xp11 translocation renal cell carcinoma
t(6;11) renal cell carcinoma
Carcinoma associated with neuroblastoma
Mucinous tubular and spindle cell carcinoma
Tubulocystic renal cell carcinoma
Acquired cystic disease associated renal cell carcinoma
Clear cell (tubulo) papillary renal cell carcinoma
Hereditary leiomyomatosis renal cell carcinoma syndrome-associated renal cell carcinoma
Renal cell carcinoma, unclassified
Metanephric tumors
Metanephric adenoma
Metanephric adenofibroma
Metanephric stromal tumor
Nephroblastic tumors
Nephrogenic rests
Nephroblastoma
Cystic partially differentiated nephroblastoma
Mesenchymal tumors occurring mainly in children
Clear cell sarcoma
Rhabdoid tumor
Congenital mesoblastic nephroma
Ossifying renal tumor of infants
Mesenchymal tumors occurring mainly in adults
Leiomyosarcoma (including renal vein)
Angiosarcoma
Rhabdomyosarcoma
Malignant fibrous histiocytoma
Hemangiopericytoma
Osteosarcoma
Synovial sarcoma
Angiomyolipoma
Epithelioid angiomyolipoma
Leiomyoma
Hemangioma
Lymphangioma
Juxtaglomerular cell tumor
Renomedullary interstitial cell tumor
Schwannoma
Solitary fibrous tumor
Mixed mesenchymal and epithelial tumors
Cystic nephroma/mixed epithelial stromal tumor
Neuroendocrine tumors
Carcinoid (low-grade neuroendocrine tumor)
Neuroendocrine carcinoma (high-grade neuroendocrine tumor)
Primitive neuroectodermal tumor
Neuroblastoma
Pheochromocytoma
Hematopoietic and lymphoid tumors
Lymphoma
Leukemia
Plasmacytoma
Germ cell tumors
Teratoma
Choriocarcinoma
Metastatic tumors
Other tumors
Table 1.2
Proposed New Renal Epithelial Tumors and emerging Tumor Entities
New epithelial tumors
Tubulocystic renal cell carcinoma
Acquired cystic disease associated renal cell carcinoma
Clear cell (tubulo) papillary renal cell carcinoma
MiT family translocation renal cell carcinoma including t(6;11) renal cell carcinoma
Hereditary leiomyomatosis renal cell carcinoma syndrome associated renal cell carcinoma
Emerging tumor entities
Thyroid-like follicular renal cell carcinoma
Succinic dehydrogenase B deficiency associated renal cell carcinoma
ALK-translocation renal cell carcinoma
Other
Renal cell carcinoma with smooth muscle stroma
Tuberous Sclerosis-associated Renal Cell Carcinoma
Rare new entities and morphologic variants of common categories have recently been described and represent important diagnostic challenges in daily practice.
1.3 Familial Renal Cancer
Hereditary renal cancers show a tendency to be multiple and bilateral, may have a family history, and present at an earlier age. Known inherited syndromes that predispose to renal tumors are listed in Table 1.3.
Table 1.3
Familial renal tumors
Syndrome
Gene
Tumor
Von Hippel-Lindau (VHL)
VHL (3p25)
Clear cell
Tuberous Sclerosis
TSC1, TSC2
Angiomyolipoma, clear cell, other
Familial renal carcinoma
Gene not identified
Clear cell
Constitutional chromosome3 translocation
Responsible gene not founda
Clear cell
Hereditary PRCC
c-MET
Papillary type 1
Birt-Hogg-Dube (BHD)
BHD
Chromophobeb
Familial oncocytoma
Loss of multiple chromosomes
Oncocytoma
Hereditary leiomyomatosis RCC
FH
Papillary type 2
1.3.1 Von Hippel-Lindau (VHL) Clear Cell Renal Cell Carcinoma (RCC)
Like its sporadic counterpart, VHL clear cell RCC harbors defective VHL tumor suppressor genes. Genetic alteration in the VHL gene in the tumor can include deletion, nonsense or frame-shift mutations, mis-sense mutations or methylation.
1.3.2 Hereditary Papillary RCC
Hereditary papillary RCC are typically bilateral, multifocal type 1 papillary RCC. Genetic alterations involve a proto-oncogene, c-MET, located at 7q31.1. Similar to what is found in sporadic papillary renal cell carcinoma, trisomy 7 and 17 are identified.
1.3.3 Hereditary Leiomyomatosis RCC
Hereditary leiomyomatosis associated-RCC patients develop cutaneous and uterine leiomyomas and one third of patients develop RCC. It was considered as a variant of type 2 papillary RCC. The pathologic findings in this disease are caused by germline mutations in the fumarate hydratase gene located at 1q42.
Architectural patterns are papillary, tubulo-papillary, tubular, solid or mixed. The morphologic hallmark of the hereditary leiomyomatosis-RCC, is the presence of large nucleus with a very prominent eosinophilic nucleolus, surrounded by a clear halo. These tumors are associated with poor prognosis.
It is now recognized a unique morphotype of RCC. The renal tumors have a papillary, alveolar, solid or tubular architecture with the characteristic features being a large nucleus and a prominent nucleolus, with a clear peripheral halo These tumors behave aggressively and have a poor prognosis when compared to other hereditary forms of RCC, or clear cell and papillary RCC (Fig. 1.1).
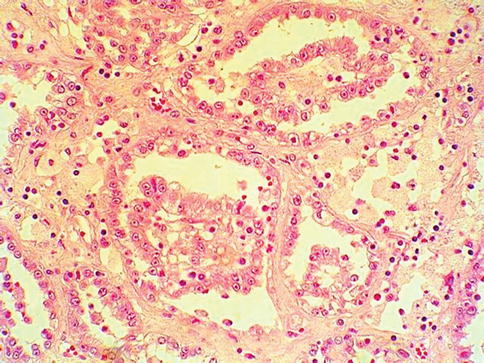
Fig. 1.1
Hereditary leiomyomatosis associated renal cell carcinoma
HLRCC leiomyomas frequently have increased cellularity, multinucleated cells, and atypia. All cases show tumor nuclei with large orangeophilic nucleoli surrounded by a perinucleolar halo similar to the changes found in HLRCC. Occasional mitoses may be seen; however, the tumors did not fulfill the criteria for malignancy.
1.3.4 Birt-Hogg-Dube Syndrome
Birt-Hogg-Dubé is an autosomal dominant cancer syndrome characterized by benign skin and renal tumors, and spontaneous pneumothorax. The disease related gene has been mapped to chromosome 17p11.2.
Birt-Hogg-Dubé is characterized by a spectrum of mutations, and clinical heterogeneity among and within families. Renal epithelial tumors with hybrid features are seen in this syndrome (Figs. 1.2 and 1.3).
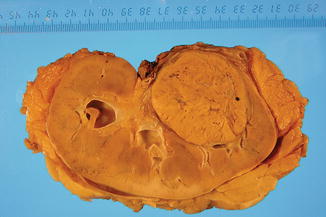
Fig. 1.2
Gross features in RCC associated with Birt-Hogg-Dubé syndrome
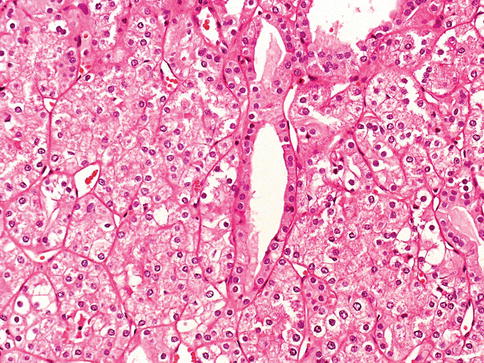
Fig. 1.3
Hybrid RCC with oncocytic and clear cells associated with Birt-Hogg-Dubé syndrome
1.4 Renal Cell Carcinoma
1.4.1 Clear Cell RCC
Most clear cell RCC are variably sized solitary cortical neoplasms, rarely bilateral (<5 %) or multicentric (4 %), typically golden yellow. Necrosis, cystic degeneration, hemorrhage, calcification, ossification, and extension into the renal vein may occur. Clear cell tumors of any size are considered malignant.
Microvascular invasion and microscopic tumor coagulative necrosis may be relevant predictors in low stage RCC (Table 1.4). Clear cell RCC has a worse prognosis when compared with chromophobe or papillary subtypes, and may progress into a sarcomatoid carcinoma which is an ominous prognostic sign (Figs. 1.4 and 1.5).
Table 1.4
TNM classification of renal cell carcinoma (2010)
PrimaryTumor (T)
TX
Primary tumor cannot be assessed
T0
No evidence of primary tumor
T1
Tumor ≤7 cm in greatest dimension, limited to the kidney
T1a
Tumor ≤4 cm in greatest dimension, limited to the kidney
T1b
Tumor >4 cm but ≤7 cm in greatest dimension, limited to the kidney
T2
Tumor >7 cm in greatest dimension, limited to the kidney
T2a
Tumor >7 cm but ≤10 cm in greatest dimension, limited to the kidney
T2b
Tumor >10 cm, limited to the kidney
T3
Tumor extends into major veins or perinephric tissues but not into the ipsilateral adrenal gland and not beyond the Gerota fascia
T3a
Tumor grossly extends into the renal vein or its segmental (muscle-containing) branches, or tumor invades perirenal and/or renal sinus fat but not beyond the Gerota fascia
T3b
Tumor grossly extends into the vena cava below the diaphragm
T3c
Tumor grossly extends into the vena cava above the diaphragm or invades the wall of the vena cava
T4
Tumor invades beyond the Gerota fascia (including contiguous extension into the ipsilateral adrenal gland)
Regional lymph nodes (N)
NX
Regional lymph nodes cannot be assessed
N0
No regional lymph node metastasis
N1
Metastasis in regional lymph node(s)
Distant metastasis (M)
M0
No distant metastasis
M1
Distant metastasis
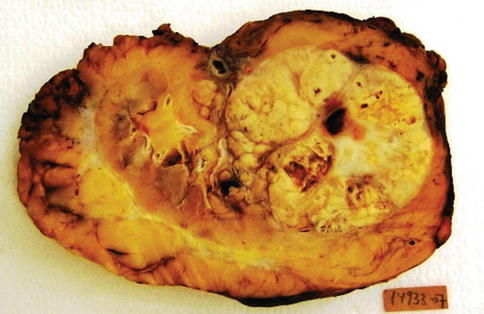
Fig. 1.4
Gross features of clear cell renal cell carcinoma
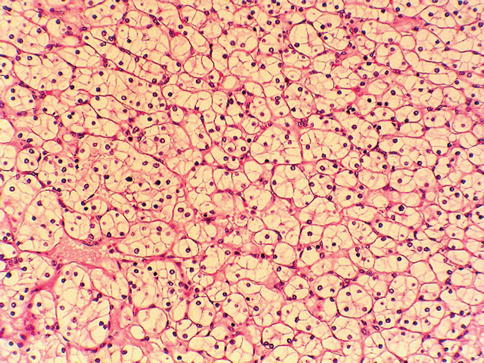
Fig. 1.5
Microscopic features of clear cell renal cell carcinoma
The international Society of Urological Pathologists (ISUP) suggested that clear cell RCC grading should be based upon nucleolar features and not Fuhrman grading (Table 1.5). Sporadic clear cell RCC displays frequent chromosome 3p losses.
Table 1.5
International Society of Urological Pathology Nucleolar Grading System
Grade
Tumor description
1
Inconspicuous or absent nucleoli at 400× magnification
2
Nucleoli distinctly visible at 400× but inconspicuous or invisible at 100× magnification
3
Nucleoli distinctly visible at 100× magnification
4
Rhabdoid or sarcomatoid differentiation, or containing tumor giant cells or showing extreme nuclear pleomorphism with clumping of chromatin
Clear cell RCC may have acidophilic cytoplasm, hemangioblastoma-like, angioleiomyoma-like stroma or pseudo-papillary architecture but retains the characteristic 3p loss.
1.4.2 Multilocular Cystic Clear Cell Renal Cell Neoplasm of Low Malignant Potential
The ISUP modification of the current 2004 WHO classification of kidney tumors recognizes multilocular cystic clear cell renal cell neoplasm of low malignant potential (MCNLMP) as a variant of clear cell RCC with a good prognosis.
This is a tumor entirely composed of cysts of variable size separated from the kidney by a fibrous capsule. The cyst are lined by a single layer of clear to pale cells but occasionally shows a few small papillae. The septa are composed of fibrous tissue that may have epithelial cells with clear cytoplasm that resemble those lining the cysts (Figs. 1.6, 1.7 and 1.8).
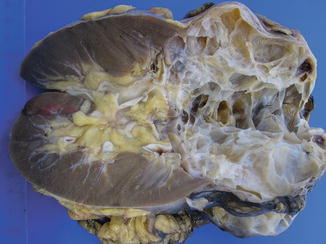
Fig. 1.6
Multilocular cystic renal cell neoplasia of low malignant potential (gross features)
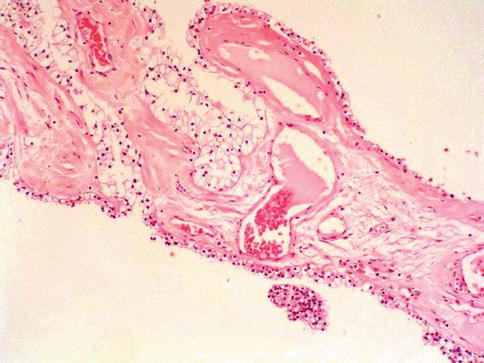
Fig. 1.7
Multilocular cystic renal cell neoplasia of low malignant potential (histologic features)
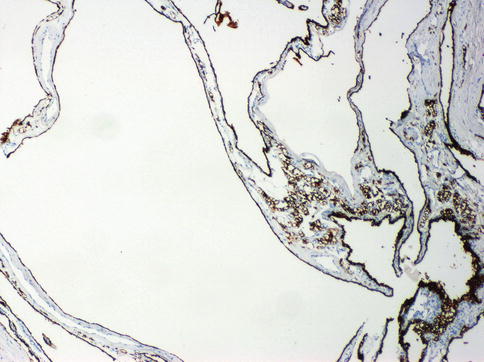
Fig. 1.8
Multilocular cystic renal cell neoplasia of low malignant potential. Expression of CAIX in the neoplastic cells
Cases with expansive nodules are excluded. VHL gene mutations in MCNLMP supports its classification as a type of clear cell RCC.
No progression of MCNLMP has been reported.
1.4.3 Papillary RCC
Papillary RCC has a less aggressive clinical course than clear cell RCC. Papillary RCC has variable proportions of papillae and may be bilateral or multifocal with hemorrhage, necrosis or cystic degeneration. The papillae contain a fibrovascular core with aggregates of foamy macrophages, calcified concretions and frequent hemosiderin granules.
Cellular type 1 and type 2 tumors have papillae covered by small cells with scanty cytoplasm arranged in a single layer in type1, and tumor cells of higher nuclear grade, eosinophilic cytoplasm and pseudostratified nuclei in type 2. Type 1 tumors have longer survival (Figs. 1.9, 1.10 and 1.11).
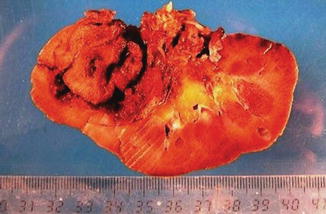
Fig. 1.9
Gross features of papillary renal cell carcinoma
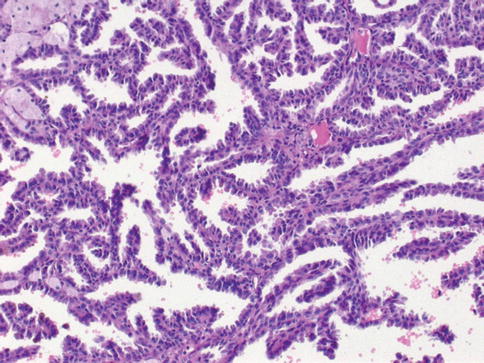
Fig. 1.10
Microscopic features of papillary carcinoma type 1
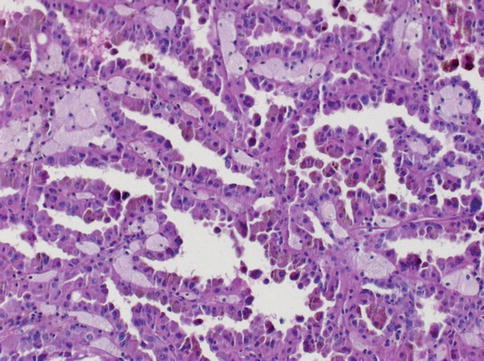
Fig. 1.11
Microscopic features of papillary carcinoma type 2
Trisomy or tetrasomy 7, trisomy 17 and loss of chromosome Y are the cytogenetic signature. Stage, tumor proliferation and sarcomatoid change being correlated with outcome. It has been suggested that papillary RCC grading should be based upon nucleolar features and not Fuhrman grading.
Age and sex distribution of papillary RCC is similar to clear cell RCC. Recent molecular genetic studies provide evidences for the independent origin of multifocal papillary tumors in patients with papillary RCC. An oncoytic variant of papillary RCC has been reported. One patient died of metastases on follow-up (Figs. 1.12 and 1.13).
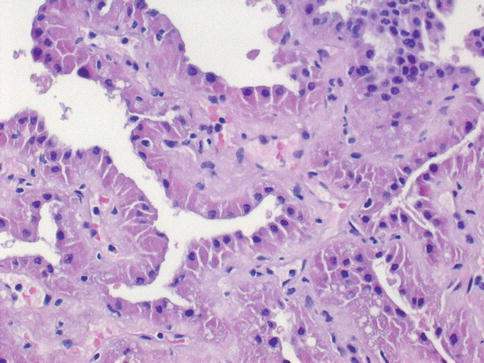
Fig. 1.12
Microscopic features of the oncocytic type of papillary carcinoma
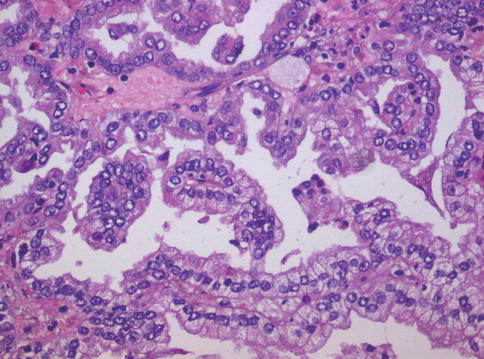
Fig. 1.13
Papillary carcinoma with focal clear cell change
1.4.4 Chromophobe RCC
Less aggressive than other RCC, the chromophobe type is characterised by huge pale cells with reticulated cytoplasm and prominent cell membrane. It accounts for 5 % of renal epithelial tumors.
Chromophobe RCC is solid and appears orange turning grey or sandy after fixation. The eosinophilic variant needs to be differentiated from oncocytoma. Sarcomatoid transformation is associated to aggressive disease. Diffuse cytoplasmic Hale’s iron colloid stain is characteristic (Figs. 1.14, 1.15 and 1.16).
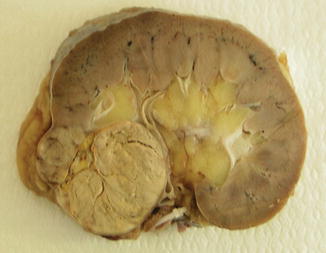
Fig. 1.14
Gross features of chromophobe renal cell carcinoma
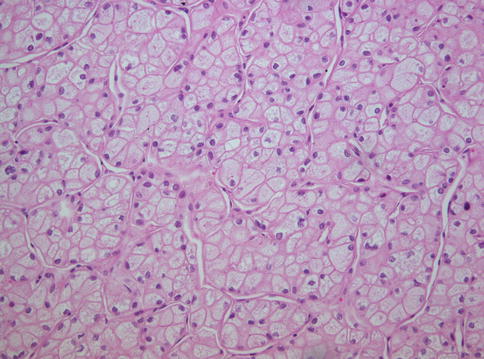
Fig. 1.15
Microscopic features of classic-type chromophobe renal cell carcinoma
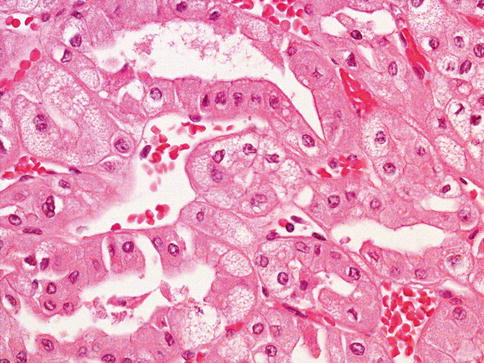
Fig. 1.16
Microscopic features of eosinophilic chromophobe renal cell carcinoma
The relationship between oncocytoma and chromophobe RCC is still unclear. Both seems to derive from the intercalated cell of the collecting duct, both have rearrangement of mitochondrial DNA, increased mitochondria in oncocytoma and numerous mitochondria-derived microvesicles in chromophobe RCC, and both are frequently observed in oncocytosis with or without Birt-Hogg-Dubé syndrome.
There are reports of hybrid tumor composed of oncocytic and chromophobe elements. Therefore, oncocytoma might be the benign counterpart of chromophobe RCC. Loss of several chromosomes characterises chromophobe RCC. Recognising occassional occurrence of metastases and 10 % mortality is of clinical relevance.
At diagnosis most patients are in the sixth decade, stage T1 or T2 (86 %) and similar gender incidence. Fuhrman grading is not appropriated to grade chromophome RCC with an international consensus on that chromophobe RCC should not be graded at present.
1.4.5 Hybrid Oncocytic Chromophobe Tumor (HOCT)
Described as tumors having a mixture of cells with the morphologic features of those seen in chromophobe RCC and renal oncocytoma (Figs. 1.17 and 1.18).
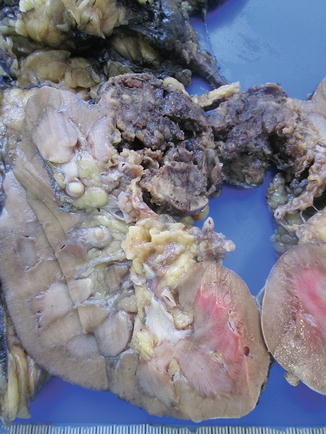
Fig. 1.17
Gross features of a case of hybrid chromophobe-oncocytoma tumor
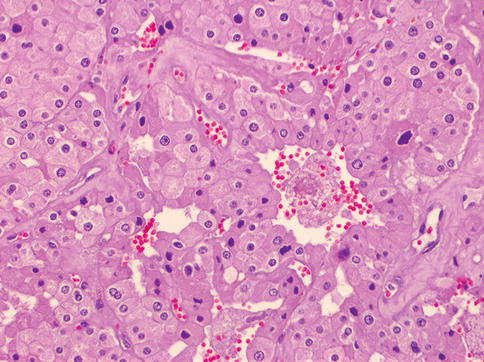
Fig. 1.18
Microscopic features of a case of hybrid chromophobe-oncocytoma tumor
HOCTs occur in three clinical situations: (i) being sporadic, (ii) in association with renal oncocytosis/oncocytomatosis, and (iii) in patients with the Birt-Hogg-Dubé syndrome (BHD). It seems that tumors from all three groups share similar morphologic features but that they have a differing molecular genetic background.
Sporadic HOCTs are composed of neoplastic cells predominantly arranged in a solid-alveolar pattern, with nuclei showing mild nuclear pleomorphism and abundant granular eosinophilic to oncocytic cytoplasm. Neoplastic cells have a perinuclear halo and are occasional binucleate. No raisinoid nuclei of the type seen in classic chromophobe RCC are present. Usually tumor cells resemble cells of oncocytoma with perinuclear cytoplasmic clearing, and, occasional, small tubules may be present.
HOCTs in oncocytosis/oncocytomatosis are almost identical to those tumors that occur sporadically, being composed of sheets of cells separated by a delicate vasculature. The cells are round to polygonal with finely granular cytoplasm, and the nuclei are slightly pleomorphic and irregular with visible nucleoli. No typical raisinoid nuclei are seen, and mitotic figures are rare.
HOCTs associated with BHD typically show three morphologic patterns: (i) an admixture of areas typical of oncocytoma and chromophobe RCC; (ii) scattered chromophobe cells in the background of a typical oncocytoma; and (iii) large eosinophilic cells with intracytoplasmic vacuoles. In these tumors, the nuclei are often more pleomorphic than other forms of HOCT and occasionally have a “raisinoid” morphology.
The majority of the tumors express parvalbumin, antimitochondrial antigen, CK7, and CD117 by immunohistochemistry. Monosomy of chromosome 20 is the most frequent genetic alteration, a finding highly unusual for both oncocytoma and chromophobe RCC.
HOCTs usually occur in adult patients, with no sex predilection; some are associated with long term hemodialysis. Can be unilateral and solitary or being bilateral and multiple lesions. Necrosis is not frequently, but central fibrotic strands/scars may be occasionally present. Tumors are usually well circumscribed and nonencapsulated with homogenous tan to brown cut surface.
HOCT, regardless of clinical association, seems to behave indolently and no evidence of aggressive behavior has been documented.
1.4.6 Collecting Duct Carcinoma
Collecting duct carcinoma (CDC), also known as Bellini’s tumor, accounts for <1 % of renal malignancies and derives from the “principal cells” of the collecting duct; ranges 2.5–12 cm, is centraly located and shows a firm grey-white appearance.
Mean patient age is 55 years with male predominance. When small, origin within a medullary pyramid may be seen at gross examination. At diagnosis, most tumors are in advanced stage with metastasis and morphologic criteria for diagnosis are the presence of an infiltrative tubular or tubulopapillary pattern, associated with desmoplastic stromal reaction, necrosis and cells displaying high Fuhrman grade (Figs. 1.19, 1.20 and 1.21).
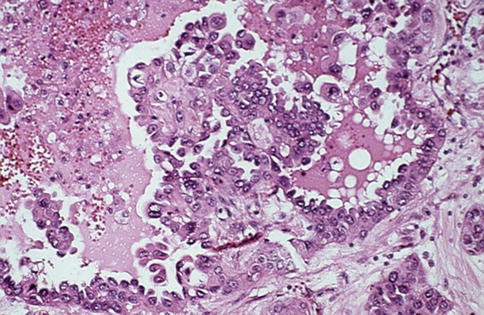
Fig. 1.19
Microscopic features of collecting duct carcinoma with papillary features
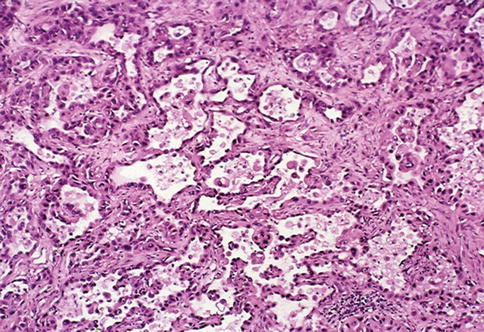
Fig. 1.20
Microscopic features of collecting duct carcinoma with anastomosing glandular channels
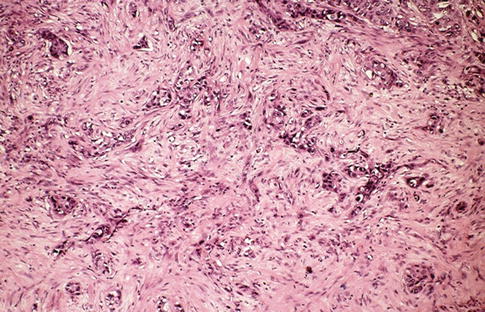
Fig. 1.21
Microscopic features of collecting duct carcinoma with desmoplastic stroma
CDC is positive for low and high molecular weight keratins, CD117 and vimentin, EMA, p63, CK7, Pax 2, Pax 8, but molecular alterations of CDC are poorly understood (Table 1.3). The main differential diagnoses of CDC include type 2 PRCC, renal pelvic adenocarcinoma or urothelial carcinoma with glandular differentiation. Immunohistochemistry is a valuable adjunct in this setting (Table 1.6).
Table 1.6
Main histotypes of renal cell tumors seen in adults with associated genetic alterations
Malignant renal cell tumors
Main genetic alterations
Clear cell renal cell carcinoma
−3p, +5q22, −6q, −8p, −9p, −14q
Multilocular cystic renal cell neoplasm of low malignant potential
VHL gene mutation
Papillary renal cell carcinoma
+3q, +7, +8, +12, +16, +17, +20, −Y
Chromophobe renal cell carcinoma
−1, −2, −6, −10, −17, −21, hypodiploidy
Carcinoma of the collecting ducts of Bellini
−1q, −6p, −8p, −13q, −21q, −3p (rare)
Tubulocystic carcinoma
Variable trisomy of chromosome 17
Renal medullary carcinoma
Rare loss of chromosome 22
MiT family translocation RCC (Renal carcinoma associated with Xp11.2 translocations/TFE3 gene fusions)
t(X;1)(p11.2;q21), t(X;17)(p11.2;q25), t(X;1)(p11.2;p34), t(X;17)(p11.2;q23), others
Renal cell carcinoma in long term survivors after neuroblastoma
Allelic imbalance at 20q13
Mucinous tubular and spindle cell carcinoma
−1, −4, −6, −8, −13, −14, +7, +11, +16, +17
Renal cell carcinoma unclassified
Variable, unknown
Acquired cystic disease-related RCC
Variable gains chromosomes 7 and 17, no VHL gene deletions, rare gains 1,2,6, 10
Clear cell papillary RCC
Lacked the gains of chromosome 7, no loss of Y chromosome, lack 3p deletions
Benign renal cell tumors
Papillary adenoma
Similar to papillary RCC but less extensive
Oncocytoma
Chromosomes 1 and/or 14 loss and frequent alterations of mitochondrial DNA, 11q13 translocation, no chromosome 3p loss
Metanephric tumors: adenoma, adenofibroma, stromal metanephric
Normal karyotypes, 2p deletion, others
Mixed stromal and epithelial tumors
Cystic nephroma/mixed epithelial and stromal tumor
Nonrandom X chromosome
Inactivation, others
One recent experience identified complete loss of expression of INI1 in 15 % and focal/weak expression of INI1 in up to 30 % of CDC, with no difference in survival by INI1 status.
1.4.7 Renal Medullary Carcinoma
It is a rapidly growing rare tumor of the renal medulla regarded as an aggressive variant of collecting duct carcinoma that was initially considered of renal pelvis origin. Some may have solid, spindled or rhabdoid phenotype (Figs. 1.22 and 1.23).
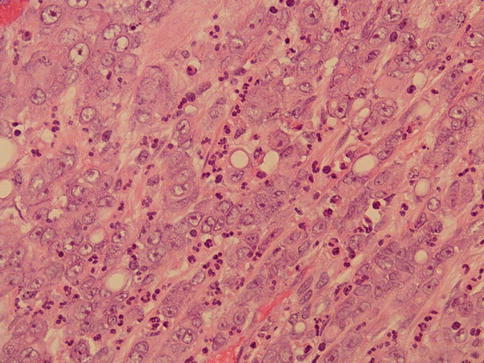
Fig. 1.22
Microscopic appearance of medullary carcinoma with associated inflammation
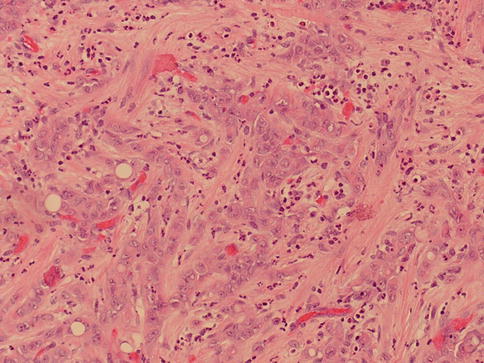
Fig. 1.23
Microscopic appearance of medullary carcinoma with desmoplasia
Immunohistochemistry analysis shows that the neoplastic cells are positive for CEA (7/8), AE1/3 (8/8), CAM5.2 (7/7), CK7 (5/5), CK20 (4/6), and vimentin (6/6)
Essentially all cases of RMC show loss of nuclear expression of the transcriptional regulator INI1/SMARCB1, reflecting prevalent mutation of the gene.
With few exceptions this tumor is seen in young male blacks with sickle cell trait (mean age 22 years), presenting with hematuria, flank pain, weight loss and palpable mass.
Metastatic disease may be the initial clinical evidence and the reported prognosis is poor.
1.4.8 Mucinous, Tubular and Spindle Cell Carcinoma
Low-grade carcinoma composed of tightly packed tubules separated by pale mucinous stroma and a spindle cell component. It seems to derive from the distal nephron (Figs. 1.24, 1.25, 1.26, 1.27 and 1.28).
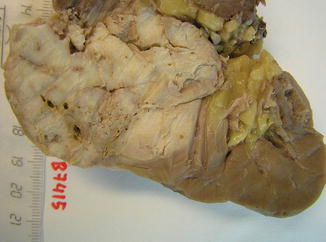
Fig. 1.24
Gross features of mucinous, spindle and tubular tumor
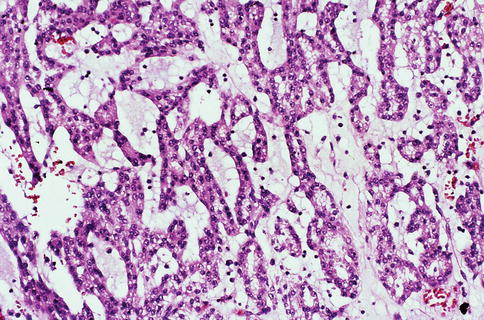
Fig. 1.25
Microscopic features of mucinous, spindle and tubular tumor (mucinous)
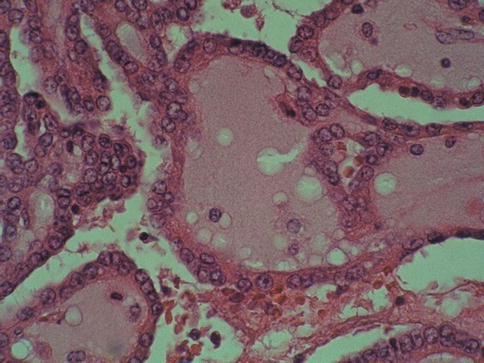
Fig. 1.26
Microscopic features of mucinous, spindle and tubular tumor (tubular)
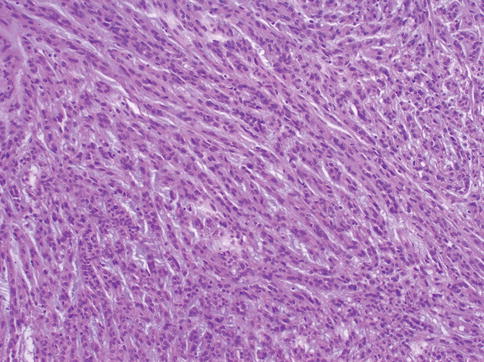
Fig. 1.27
Microscopic features of mucinous, spindle and tubular tumor (spindle)
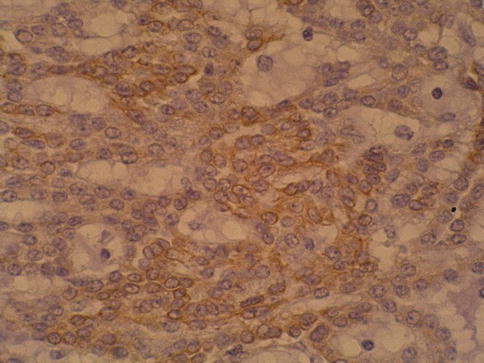
Fig. 1.28
Focal positivity of mucinous, spindle and tubular tumor with 34BE12
This tumor has a combination of losses involving chromosomes 1, 4, 6, 8, 13 and 14 and gains of chromosome 7, 11, 16 and 17. Immunohistochemical analysis found a significant overlap with papillary RCC, and some authors believe this is a variant of papillary RCC with spindle cell differentiation.
There is a female predominance and the mean age is 50 years at diagnosis. One patient developed metastases on follow-up.
1.4.9 Renal Cell Carcinoma After Neuroblastoma
A few cases of RCC arise in long term survivors of childhood neuroblastoma. This group is heterogeneous that shows oncocytoid features.
Allelic imbalances occur at the 20q13 locus. The prognosis is similar to other RCC. Uni- or bilateral lesions develop at mean age of 13 years.
1.4.10 RCC with Sarcomatoid or Rhabdoid Differentiation
Current WHO classification does not consider sarcomatoid RCC as an entity but rather as a progression of any RCC main type. Pure sarcomatoid morphology without recognizable epithelial elements falls into the unclassified RCC category (Figs. 1.29 and 1.30).
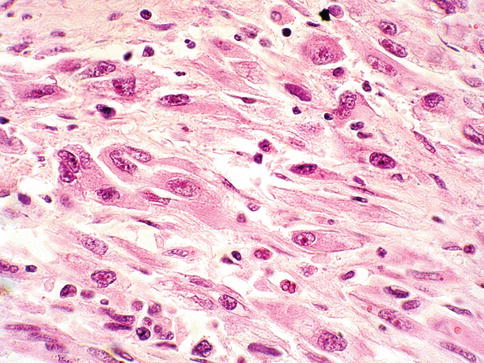
Fig. 1.29
Sarcomatoid renal cell carcinoma
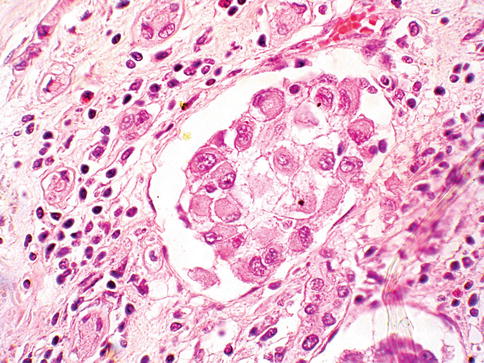
Fig. 1.30
Renal cell carcinoma with rhabdoid features
RCC with sarcomatoid elements show higher proliferative activity than other renal cell carcinoma types and usually exhibit highly malignant behavior with a predilection for increased local invasiveness and a higher likelihood of distant metastasis.
Sarcomatoid components may be seen in clear cell, papillary, chromophobe and collecting duct carcinomas. It is speculated that the sarcomatoid components of RCC represent areas of dedifferentiation or epithelial-mesenchymal transition.
Patterns of allelic loss and of nonrandom X-chromosome inactivation in clear cell and sarcomatoid components of RCC from 22 patients and concluded that both clear cell and sarcomatoid components of RCC are derived from the same progenitor cell.
The specific molecular mechanisms responsible for sarcomatoid transformation of a renal tumor are unknown, although some studies suggest a link with mutations of the TP53 tumor suppressor gene.
RCC with rhabdoid differentiation is a rare and aggressive neoplasm with poor prognosis. Most patients are at high stage at diagnosis, develop metastases soon after and died of the disease within a year of diagnosis.
Rhabdoid cells are large with eccentric atypical nucleus and eosinophilic intracytoplasmic inclusion that is positive for vimentin, EMA, and cytokeratin. Sarcomatoid change and rhabdoid features may coexist. Rhabdoid cells show loss of nuclear expression of the transcriptional regulator INI1/SMARCB1
1.4.11 Renal Cell Carcinoma, Unclassified
It represents 4–6 % of renal tumors. At diagnosis, most are of high grade and stage with poor survival (Figs. 1.31 and 1.32).
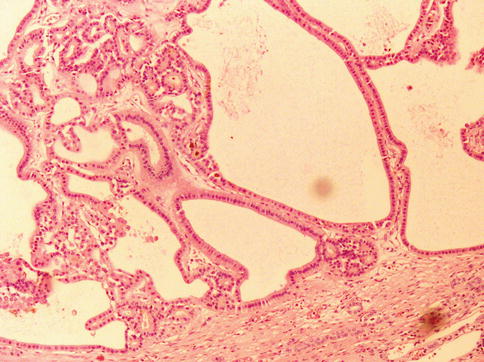
Fig. 1.31
Renal cell carcinoma unclassified with variable sized tubules
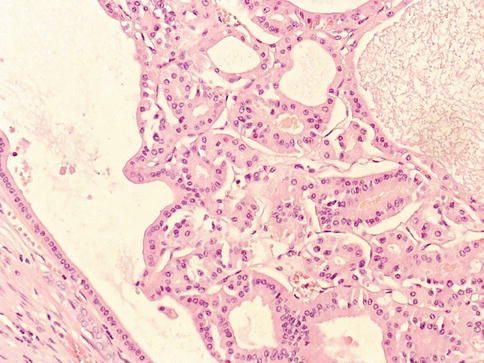
Fig. 1.32
Renal cell carcinoma unclassified with small sized tubules
The WHO criteria include: (i) composites of recognised types, (ii) pure sarcomatoid morphology, (iii) mucin production, (iv) rare mixtures of epithelial and stromal elements, and (v) unrecognisable cell types.
Reported data suggests that it is an aggressive form of RCC as confirmed in a recent study based on 56 cases. The prognosis of these patients is mainly related to pT stage, tumor size, vascular invasion, tumor necrosis or recurrence after surgery.
1.5 Proposed New and Emerging Epithelial Renal Tumors
1.5.1 Tubulocystic Renal Cell Carcinoma
Is a rare renal tumor composed of tubular and cystic structures. The genomic alterations of tubulocystic carcinoma are alike but not identical to those of papillary RCC (Fig. 1.33).
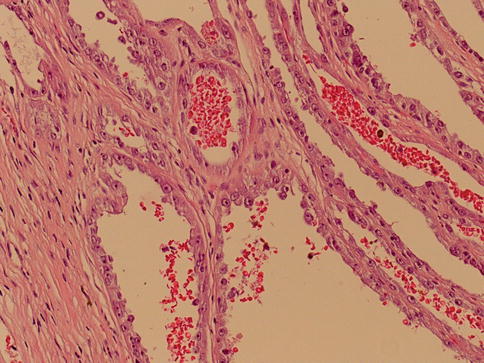
Fig. 1.33
Tubulocystic renal cell carcinoma
Like papillary RCC, it often exhibits trisomy of chromosome 17, but it does not show trisomy 7. It does not exhibit monosomy of chromosomes 1, 6, 14, 15, and 22 and frequent allelic loss on chromosomal arms 1q, 6p, 8p, 13q, and 21q, which are frequently seen in collecting duct cancer.
Immunohistochemistry showed variable expression of CD10, AMACR, parvalbumin, 34βE12, PAX-2, CAIX, CK8, 18, CK19 and occasionally for CK7 and 34βE12.
Rare cases may present with lymph node metastasis. Some may coexist with papillary RCC.
1.5.2 Acquired Cystic Disease-Associated RCC
These are composed of cells with abundant eosinophilic cytoplasm and variably solid, cribriform, tubulocystic and papillary architecture. It shows no VHL gene deletions, although gains of chromosomes 7 and 17 are present in some cases.
Deposits of calcium oxalate crystals are frequent finding in each tumor. One study based on FISH analysis showed no losses or gains of chromosomes 1, 2, 6, 10, or 17 in one tumor; gains of chromosomes 1, 2, and 6 were noted in two tumors, one of these also showed gains of chromosome 10 (Fig. 1.34).
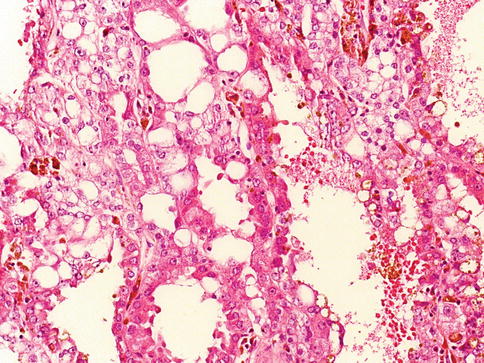
Fig. 1.34
Acquired cystic disease related renal cell carcinoma
1.5.3 Clear Cell Papillary (Tubulo-Papillary) RCC
These are renal carcinomas composed mainly of papillary structures proliferating within cystic spaces, lined by cells with clear cytoplasm.
All tumors lacked the gains of chromosome 7 and loss of Y that are typical for papillary renal cell carcinoma and furthermore lack the 3p deletion which is typical of clear cell RCC (Figs. 1.35 and 1.36).
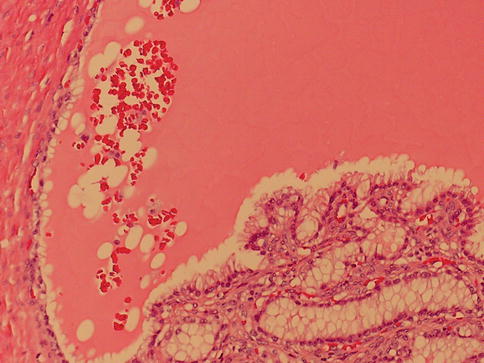
Fig. 1.35
Clear cell papillary renal cell carcinoma, cystic form
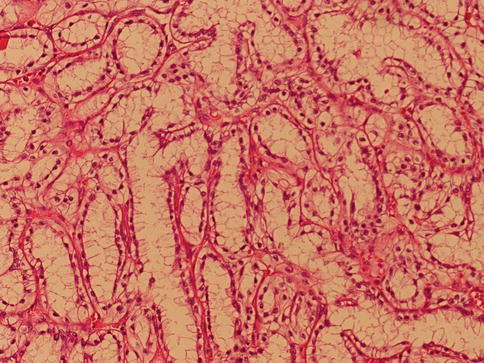
Fig. 1.36
Non-cystic clear cell papillary renal cell carcinoma
These tumors also occur in patients unrelated to end-stage renal disease.
1.5.4 MiT Family Translocation RCC
This type of RCC is defined by different translocations involving chromosome Xp11.2, all resulting in gene fusions involving the TFE3 gene. This carcinoma predominantly affects children and young adults, but may be seen in adults.
The ASPL-TFE3 translocation carcinomas characteristically present at an advanced stage associated with lymph node metastases. RCC associated with Xp11.2 translocations resemble clear cell RCC on gross examination and seems to have an indolent evolution, even with metastasis (Figs. 1.37 and 1.38).
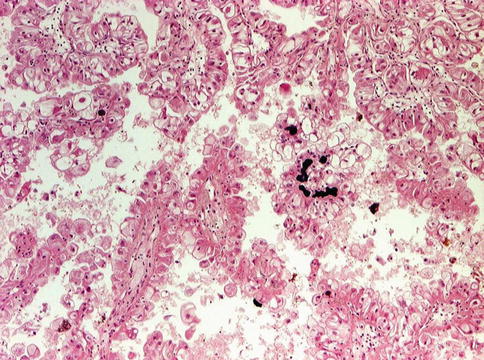
Fig. 1.37
TFE3 translocation carcinoma with papillary architecture and calcification
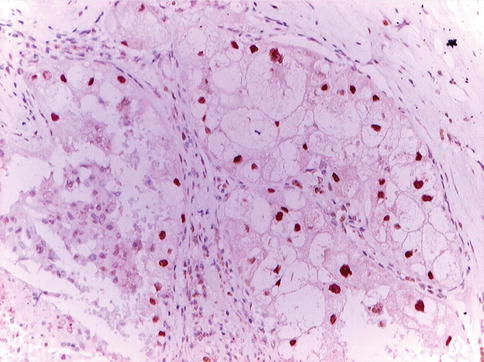
Fig. 1.38
TFE3 positive immunohistochemistry in translocation carcinoma
The histopathologic appearance is that of a papillary carcinoma with clear cells and cells with granular eosinophilic cytoplasm with foci of calcifications regardless of the type of translocation.
TFE3 immunostainings were positive in only 82 % of TFE3 translocation carcinomas. Both TFE3 and TFEB renal translocation carcinomas expressed CD10 and alpha-methylacyl-coenzyme-A racemase.
Another subset of renal tumors are associated with a translocation t(6;11)(p21;q12) involving the transcription factor EB (TFEB). Translocation involving TFE3 and TFEB can be specifically identified by immunohistochemistry, but diagnosis may also be performed by FISH analysis (Fig. 1.39).
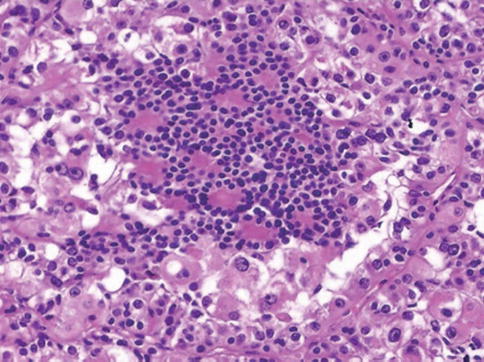
Fig. 1.39
TFEB associated translocation carcinoma
Labeling for PAX8 distinguishes the t(6;11) RCC from epithelioid angiomyolipoma, which otherwise shares a similar immunoprofile.
1.5.5 Thyroid-Like Follicular Renal Cell Carcinoma
It is a rare variant of RCC resembling well differentiated follicular carcinoma of the thyroid gland. These tumors are well circumscribed, tan brown and homogenous.
Microscopically they consist of tumor follicles of varying size containing dense eosinophilic colloid-like material (Fig. 1.40). The genetics and immunohistochemical expression of these tumors are, as yet, not fully characterized although thyroid follicular cell markers, thyroglobulin and TTF-1 are negative.
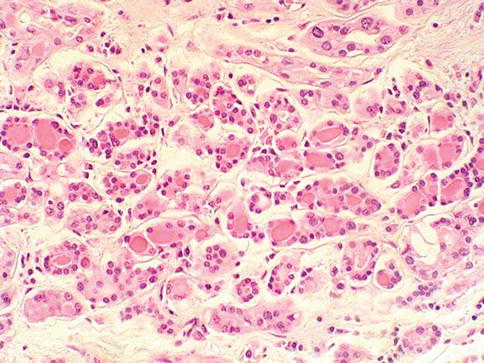
Fig. 1.40
Thyroid-like follicular renal cell carcinoma
The majority of cases reported to date have shown an indolent behavior, although metastases to lymph nodes in two cases and a single case with pulmonary metastases have been reported.
1.5.6 Succinic Dehydrogenase B Deficiency Associated Renal Cell Carcinoma
These tumors are seen in patients within germline Succinate Dehydrogenase B mutation, which is associated with a syndrome characterized by pheochromocytoma, paraganglioma and type 2 gastrointestinal stromal tumors.
Microscopically these tumors are characterized by the presence of compact nests of eosinophilic polygonal cells. Cytoplasmic vacuoles and pale eosinophilic cytoplasmic inclusions are commonly seen and these represent giant mitochondria.
Less than ten cases have been reported to date and follow-up is limited. Of note, sarcomatoid change was identified in two patients with metastatic disease, one of whom died of tumor related causes.
1.5.7 ALK-Translocation Renal Cell Carcinoma
A fusion of the anaplastic lymphoma kinase (ALK) gene with the gene for the cytoskeletal protein vinculin (VCL) resulting from the translocation t(2:10)(p23;q22) has been reported in two tumors observed in young patients.
VCL–ALK translocation RCC appear to have a characteristic appearance, consisting of cells with a polygonal to spindled morphology with abundant eosinophilic cytoplasm and frequent intracytoplasmic lumina.
While these two cases were associated with sickle cell trait, two further RCCs showing ALK fusion with different partner genes have been described in older patients, although neither of these showed a sickle cell association.
Two additional cases of RCC demonstrating ALK fusion (fusion partner unknown) have been reported in adult patients that behave aggressively.
A recent case showing VCL–ALK RCC in a child with sickle-cell trait has been recently reported.< div class='tao-gold-member'>Only gold members can continue reading. Log In or Register to continue
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


