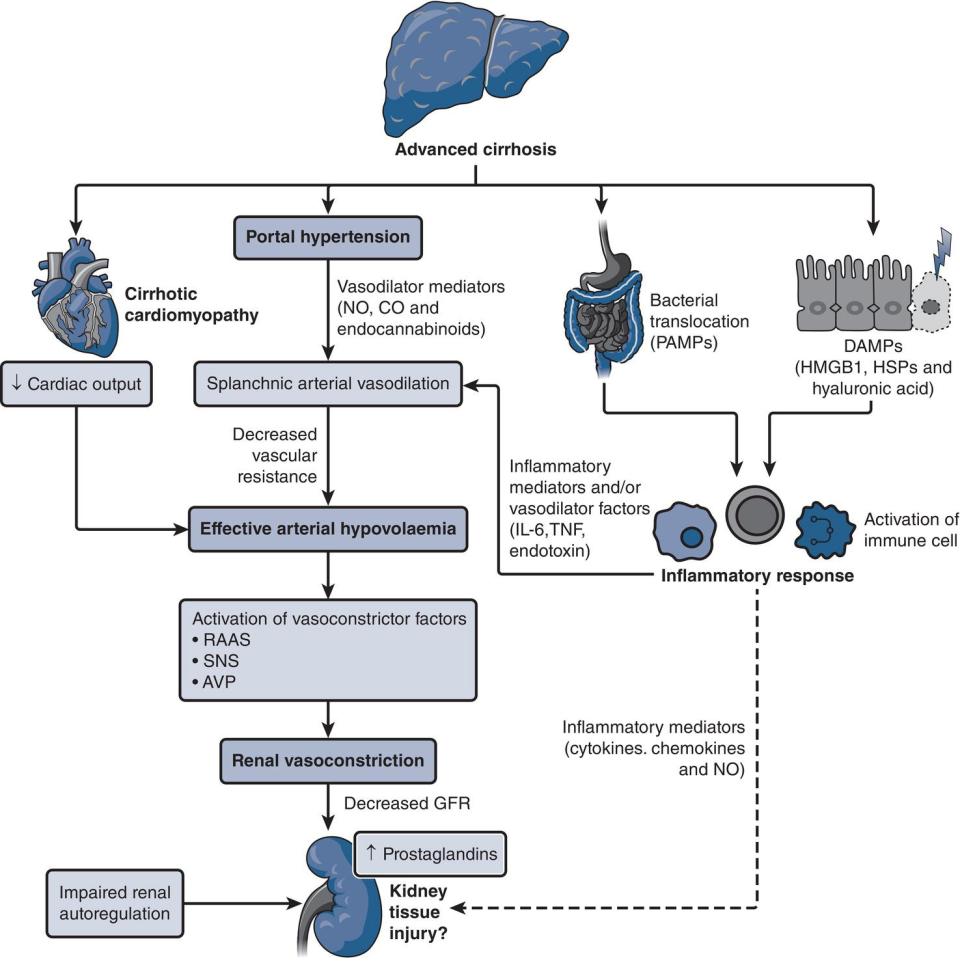
Javeria Peracha and Graham Lipkin Queen Elizabeth Hospital, Birmingham, UK Patients with abnormal liver tests are commonly encountered by renal physicians across the breadth of clinical practice. Hepatic and renal function are closely intertwined through involvement in systemic disease processes, hemodynamic interrelationships, coexisting primary organ disorders, and drug toxicity. This chapter starts by examining factors related to the assessment and management of AKI in patients with liver cirrhosis, one of the most frequently encountered scenarios in clinical practice. CKD in patients with cirrhosis is then be explored, alongside renal impairment in the context of acute liver failure (ALF) and liver transplantation. We then review common causes of abnormal elevations in liver enzymes among patients with renal disease, including AKI, CKD (including dialysis), and renal transplantation. Each section in this chapter outlines key differential diagnoses relevant to the clinical settings and a framework that will allow further investigation and assessment of these conditions. We briefly touch on pathophysiology and management of some key conditions, including HRS, and the challenges surrounding viral hepatitis screening, diagnosis, and management amongst patients with AKI, CKD (including dialysis), and renal transplants. Limitations of creatinine‐based estimates of kidney function in patients with cirrhosis including calculated estimated glomerular filtration rate (eGFR) should be recognized, due to sarcopenia, altered tubular secretion of creatinine, and an increased volume of distribution (Box 12.1). Serum cystatin C level may provide a more sensitive and reliable marker of renal function in patients with cirrhosis, although laboratory measurement is not widely available. When accurate measurement of glomerular filtration rate (GFR) is required (e.g. for transplant assessment), isotopic clearance of 99Tc DTPA (diethylenetriaminepentaacetic acid) or iohexol overcomes some of the inaccuracies associated with creatinine‐based measurement [1]. AKI describes a sudden reduction in kidney function, observed in up to 50% of hospitalized patients with cirrhosis and is associated with substantial additional morbidity and mortality. The International Ascites Club standardized AKI diagnostic criteria for patients with cirrhosis is based on the Kidney Disease: Improving Global Outcomes (KDIGO) AKI clinical practice guidelines (Table 12.1) [2]. However, trends in serum creatinine predominate over urine output parameters, which can be unreliable in patients with cirrhosis, where avid sodium and water retention can cause oliguria, independent of true kidney function [2, 3]. Most cases of AKI in patients with cirrhosis are “pre‐renal,” due to reduced glomerular perfusion on top of reduced renal blood flow. This is frequently precipitated by infection (46%), including bacterial peritonitis or relative hypovolemia (32%), such as when there is variceal bleeding or paracentesis without adequate albumin replacement (Table 12.2). Prolonged or severe pre‐renal AKI may progress to acute tubular necrosis (ATN). Less commonly, primary renal parenchymal disease may cause AKI. Associations of liver disease with glomerulonephritis are outlined in Table 12.3. Bile‐cast nephropathy leading to AKI or a Fanconi‐like proximal tubulopathy (characterized by low serum uric acid and phosphate) is associated with severe hyperbilirubinemia. Particular attention should be paid to management of medication as patients with cirrhosis are at risk from nephrotoxic medications. Table 12.1 International Ascites Club acute kidney injury diagnostic criteria. AKI, acute kidney injury; SCr, serum creatinine. a Stage 1 is divided further into stage 1a (sCr < 132.6 μmol/l or 1.5 mg/dl) and 1b (sCr > 132.6 μmol/l or 1.5 mg/dl) [2]. Table 12.2 Etiology of acute kidney injury in patients hospitalized with liver cirrhosis. Adapted from [4]. Table 12.3 Glomerulonephritis reported in association with chronic liver disease. “AKI ”, previously referred to as “type 1 HRS,” is a diagnosis of exclusion, resulting from the renal physiological changes seen in advanced liver cirrhosis, in the absence of structural or parenchymal renal disease (Figure 12.1). Development of portal hypertension leads to increased release of vasodilatory mediators into the splanchnic arterial circulation (nitric oxide, prostacyclin, carbon monoxide, and endogenous cannabinoids). Pooling of blood within the splanchnic system causes “effective arterial hypovolemia” and reduced renal perfusion. The simultaneous release of damage‐associated molecular patterns from cirrhotic liver tissue and pathogen‐associated molecular patterns from bacterial translocation in the gut are thought to activate circulating innate immune cells, triggering a systemic inflammatory response and release of cytokines (including tumor necrosis factor alpha and interleukin‐6) into the circulation, which may further exacerbate circulatory dysfunction. Figure 12.1 Pathophysiology of acute kidney injury associated with hepatorenal syndrome. AVP, arginine vasopressin; DAMPs, damage‐associated molecular patterns; GFR, glomerular filtration rate; HMGB1, High mobility group box protein 1; HSPs, heat shock proteins; LI‐6, interleukin‐6; NO, nitric oxide; PAMPs, pathogen‐associated molecular patterns; RAAS, renin–angiotensin–aldosterone system; SNS, sympathetic nervous system; TNF, tumor necrosis factor. In the earlier stages of chronic liver disease, a compensatory increase in cardiac output helps to maintain adequate renal perfusion. As hepatic dysfunction progresses, however, this compensatory mechanism is overwhelmed, exacerbated by the presence of “cirrhotic cardiomyopathy” (characterized by systolic and diastolic dysfunction, seen in up to 50% of patients with cirrhosis). Compensatory activation of the sympathetic nervous system, the renin–angiotensin–aldosterone system, and vasopressin release in addition may lead to intense renal vasoconstriction and a decline in GFR, alongside increased sodium and water retention, contributing to ascites formation. Initial evaluation to determine the underlying cause of AKI should include a detailed history and examination, including review of the drug treatment chart. Important investigations include: In patients where the cause of AKI remains unclear or if patients are slow to respond to initial resuscitation, the following additional investigations are suggested: Figure 12.2 Urine microscopy. Source: Reilly 2013 / with permission of McGraw Hill. Usatine 2013 / with permission of McGraw‐Hill. Mohapatra 2016 / Oxford University Press. A step‐by‐step approach to management of AKI in patients with liver disease is outlined in (Figure 12.4). For patients with AKI, it is important stop any nephrotoxins such as non‐steroidal anti‐inflammatory drugs, aminoglycosides, or angiotensin‐converting enzyme inhibitors/angiotensin receptor blockers. Vasodilator agents and laxatives should be discontinued in patients with severe diarrhea. Where intravascular volume depletion is present, diuretics should be withheld, and cautious fluid resuscitation, initiated with crystalloids or blood products (if hemoglobin < 70 g/l), followed by volume expansion with albumin (1 g/kg/day for 48 hours) if renal impairment persists. There should be a low threshold for commencing antibiotic therapy in cases of suspected infection. Assessment to guide continuing resuscitation is notoriously difficult, requiring regular clinical review in the setting of a high‐dependency environment. Strict fluid balance, daily weight and measurement of postural blood pressure, hourly urine output, and central venous pressure may facilitate this process. Figure 12.3 Cutaneous cryoglobulinemic vasculitis. Palpable purpura of the legs with (i) isolated, (ii) confluent lesions, and (ii) cutaneous perimalleolar ulcers. Source: Garini 2010 / with permission of Elsevier. Key diagnostic criteria for AKI‐HRS are outlined in (Box 12.3). Following albumin replacement, persistent AKI‐HRS may be treated with splanchnic vasoconstriction using, for example, terlipressin. Terlipressin has been shown to reduce mortality risk in patients with AKI‐HRS and to increase the proportion of patients who recover their renal function. Of note, terlipressin is not yet licensed for this indication in many countries, and caution is advised regarding its adverse effect profile, including severe abdominal pain and diarrhea, increased risk of cardiovascular events and respiratory failure. Alternative vasoconstrictors such as noradrenaline or midodrine/octreotide may be used, but there is increasing evidence to suggest that the latter are less effective therapies. Patients with AKI‐HRS require close monitoring to prevent fluid overload and progressive hyponatremia [5, 6], Table 12.4 Renal screen – suggested immunology blood test panel. Ig, immunoglobulin; MPO, myeloperoxidase; PR3, proteinase 3; SLE, systemic lupus erythematosus. Figure 12.4 International Club of Ascites algorithm for the management of acute kidney injury (AKI) in patients with cirrhosis. Source: Sanchez and Francoz. [3] /with Permission of John Wiley & Sons. * Return of serum creatinine to a value within 0.3 mg/ml (26.5 μmol/l) from baseline. ** Specific diseases such as acute glomerulopathies or acute vascular diseases may require specific treatments discussed with nephrologist; in contrast, there is no specific treatment for acute tubular necrosis. HRS, hepatorenal syndrome; NSAIDs, non‐steroidal anti‐inflammatory drugs.
12
Renal Medicine
Introduction
Renal Impairment in Patients with Liver Disease
Measurement of Renal Function in Patients with Cirrhosis
Acute Kidney Injury in Patients with Liver Cirrhosis
Etiology of Acute Kidney Injury
Subject
Definition
Baseline sCr
A value of sCr obtained in the previous three months
In patients with more than one sCr, the one closest to the hospital admission should be used
In patients with no previous sCr, the value on admission should be used
Definition of AKI
Increase in serum creatinine (sCr) of ≥ 0.3 mg/dl (≥ 26.5 μmol/l) within 48 hours or a > 50% rise in sCr from “baseline”
Staging:
1a
Increase in sCr ≥ 1.5 to 2 fold from baselinea
Or increase in sCr by ≥ 0.3 mg/dl (≥26.5 μmol/l) within 48 hours
2
Increase in sCr ≥ 2–3‐fold from baselinea
Increase in sCr ≥ 3‐fold from baselinea
3
Or increase in sCr to ≥4.0 mg/dl (≥353.6 μmol/l)
Or initiation of renal replacement therapy
Cause
Frequency (%)
Precipitants
Infection associated
46
Sepsis
Spontaneous bacterial peritonitis
Hypovolemia associated
32
Diuretic overuse
Gastrointestinal bleeding (including variceal bleeds)
Lactulose and infection‐related diarrhea
Large‐volume paracentesis (without adequate albumin replacement)
Hepatorenal syndrome
13
Decompensated liver cirrhosis
Parenchymal renal disease
9
Acute tubular necrosis
Interstitial nephritis
Bile‐cast nephropathy
Glomerulonephritis
Disease
Association
Alcoholic cirrhosis
Immunoglobulin A nephropathy
Hepatitis B
Membranous nephropathy (most common)
Polyarteritis nodosa
Membranoproliferative glomerulonephritis
Hepatitis C
Membranoproliferative glomerulonephritis with or without cryoglobulins (most common)
Membranous nephropathy
Fibrillary glomerulonephritis
Primary biliary cholangitis
Membranous nephropathy
Anti‐neutrophilic cytoplasmic antibody vasculitis
Autoimmune hepatitis
Immune complex glomerulonephritis
Membranous nephropathy
Membranoproliferative glomerulonephritis
Alpha‐1‐antitrypsin deficiency
Membranoproliferative glomerulonephritis
Acute Kidney Injury Hepatorenal Syndrome
Investigation

Management

Test
Associated conditions
Immunoglobulins A, G, M
IgA nephropathy, myeloma
Protein electrophoresis
Myeloma
Serum free light chains
Myeloma
ANA and dsDNA
SLE
Complement factors (C3 and C4)
SLE, cryoglobulinemic vasculitis
Anti‐neutrophil cytoplasmic antibodies MPO and PR3
Vasculitis
Anti‐glomerular basement membrane antibodies (GBM)
Goodpasture’s syndrome
Anti‐streptolysin O titer (ASOT)
Post‐infectious glomerulonephritis
Angiotensin‐converting enzyme (ACE)
Sarcoidosis
Cryoglobulins
Hepatitis C virus associated membranoproliferative glomerulonephritis
Phospholipase A2 receptor antibodies (PLA2R)
Idiopathic membranous nephropathy
Rheumatoid factor
Cryoglobulinaemic vasculitis

Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


