Protein
Mol. Wt. (kDa)
Function
Ligand binding
Albumin
66
Binding and carrier protein, osmotic regulator
Hormones, amino acids, steroids, vitamins, fatty acids
α-1-acid glycoprotein (orosomucoid)
40
Uncertain, may have role in inflammation. Acute phase reactant
α-1-antitrypsin
54
General protease inhibitor
Proteases in serum and tissue secretions
α-fetoprotein
72
Uncertain, expressed in fetus and malignancy. Tumor marker
Possibly the same as albumin
α-2-macroglobulin
720
Serum endoprotease inhibitor
Proteases
Antithrombin III
65
Protease inhibitor of intrinsic coagulation system
Proteases
Apolipoprotein A-I
26
Lecithin:cholesterolacyltransferase activator. Esterifies cholesterol in HDL
Major apolipoprotein of plasma HDL
Apolipoprotein B-100
500
Lipoprotein assembly and secretion, LDL receptor ligand
Component of plasma VLDL and LDL, binds to LDL receptor
Ceruloplasmin
134
Transport of copper
6 atoms copper/mol
C-reactive protein
105
Uncertain, may have role in inflammation. Acute phase reactant
Complement C1q
Fibrinogen
340
Fibrin precursor in hemostasis
Haptoglobin
100
Binding and transport of cell-free hemoglobin
Hemoglobin
Hemopexin
57
Binds to porphyrins, particularly heme for recycling
Porphyrins
Transferrin
80
Iron transport
2 atoms iron/mol
One especially interesting glycoprotein produced by the liver is important in both health and disease. Alpha-1-antitrypsin (A-1-AT) is a neutrophil protease inhibitor that is secreted and protects tissues, particularly lung, from damage by endogenous proteases. In A-1-AT deficiency, a point mutation produces an abnormal protein that accumulates in the ER and is susceptible to misfolding, polymerization and aggregation, resulting in toxicity to the hepatocyte. This disorder is a common cause of liver disease in infants and adults, as well as chronic lung disease in adults. Interestingly, only about 10 % of homozygotes develop clinically significant liver disease that may require liver transplantation. Genetic and environmental modifiers that impact on the adaptation to or degradation of the accumulated abnormal protein in the hepatocyte ER, through proteosomal and autophagic pathways, appear to play crucial roles in determining the severity of the liver disease. Development of new therapies has focused on targeting and enhancing these pathways.
3 Role of the Liver in Carbohydrate Metabolism
The liver plays a key role in the utilization of the major monosaccharides , glucose, fructose, and galactose (Fig. 11.1). The initial step in hepatic glucose metabolism is phosphorylation by glucokinase . Glucokinase synthesis is increased by high glucose diets and high insulin concentrations. Depending upon the metabolic need, the glucose may be utilized for energy production, synthesis of other substrates (i.e. amino acids, fatty acids), or stored as glycogen. Glycogen is a polymer of units of glucose (Fig. 11.2) with linear 1-4 linkages and branch points with 1-6 linkages. Glycogen serves as a storage depot for glucose in the liver, which can be readily mobilized when glucose is in immediate demand. Because of its polymeric structure, glycogen has a low osmolality and is more easily stored in hepatocytes than the monomeric glucose. The liver can store up to 65 g of glycogen per kilogram of liver tissue.
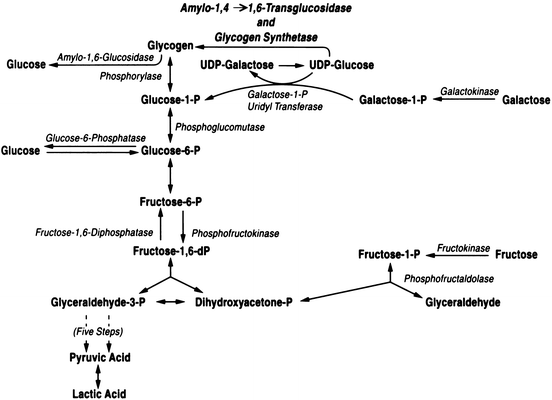
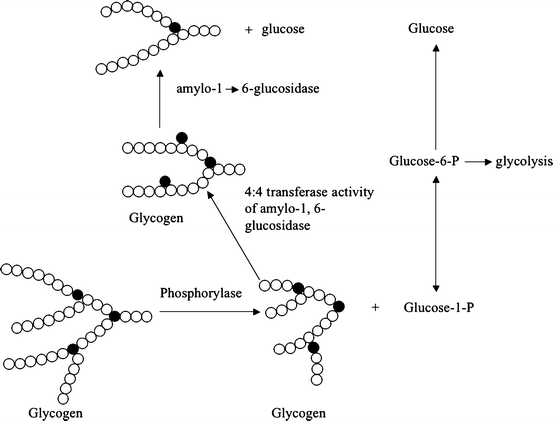
Fig. 11.1
Metabolic pathways for hepatic utilization of glucose, fructose, and galactose
Fig. 11.2
Pathway for production of glucose from glycogen (glycogenolysis). Notice that the product of the phosphorylase-catalyzed hydrolysis of glycogen is glucose-1-phosphate. A small amount of glucose (about 6 % of the total produced) is released via action of the enzyme amylo-1,6-glucosidase. This last reaction removes the glucose residues that form branch points (closed circles) and occurs after the shortened oligosaccharide chain attached to a branch point (three glucose residues) is shifted to create a longer oligosaccharide. These reactions are required because phosphorylase will not catalyze hydrolysis of short oligosaccharide chains (Reproduced with permission from Zakim [16, p. 69])
Fructose is taken up by the hepatocyte and phosphorylated by fuctokinase (Fig. 11.1). It may then be stored as glycogen by conversion to glucose-6-phosphate . When used for energy production, phosphorylated fructose may actually traverse the glycolytic pathway more readily than glucose. Also, fructose is a better substrate for lipogenesis than glucose in the liver . Hepatic metabolism of fructose follows the Leloir pathway to form either glucose-6-phosphate to enter the glycolytic pathway or UDP-glucose to enter the glycogenesis pathway. Galactose can also be taken up and used by the liver after phosphorylation by galactokinase (Fig. 11.1).
Glycolysis is the only pathway by which glucose can be oxidized anaerobically with production of ATP. Under aerobic conditions the liver uses mainly fatty acids as substrates for oxidation, and the glycolytic rate is low. Higher rates of glycolysis result in lipogenesis from carbohydrate. Regulation of glycolysis in the liver is highly integrated with that of gluconeogenesis, lipogenesis, glycogen synthesis, and glycogenolysis.
Gluconeogenesis is the production of glucose from amino acids and lactate and is carried out solely in liver and renal cortex. Two cycles exist for hepatic gluconeogenesis from non-hepatic substrates (Fig. 11.3). These are the lactic acid (Cori) cycle and the glucose-alanine cycle . In the Cori cycle lactic acid produced by working muscle is taken up by the liver and provides a substrate for gluconeogenesis to produce glucose. Alanine is also released from muscle and serves as a substrate for hepatic glucose synthesis. Regulation of gluconeogenesis is dependent on substrate availability and hormonal factors, particularly insulin and glucagon levels. Overall, glucagon and epinephrine stimulate and insulin inhibits gluconeogenesis.
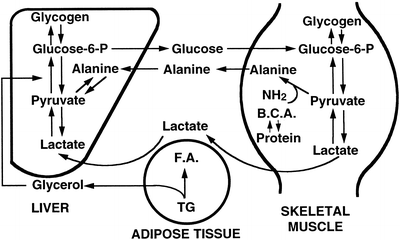
Fig. 11.3
The lactic acid (Cori) and glucose-alanine cycles. Lactic acid produced by working muscle is taken up by the liver and provides a substrate for gluconeogenesis to produce glucose. Alanine produced in muscle as a result of protein breakdown, is released and deaminated in the liver to form pyruvate. Glucose is formed from pyruvate and is released to be metabolized by muscle, where it is reconverted to pyruvate and alanine, respectively, completing the cycle. BCA branched-chain amino acids, FA fatty acid, TG triglyceride (Reproduced with permission from Van Thiel [13])
As mentioned previously, glycogen is a storage form of glucose consisting of a polymeric form of glucose. The pathway for glycogen metabolism is shown in Fig. 11.4. Most substrates enter the glycogen synthetic pathway by conversion to UDP-glucose . The glucosyl units are linked linearly by 1-4 linkages by the enzyme UDPG-glycogen synthetase. Branch points with 1-6 linkages are formed by the branching enzyme amylo-1-4, 1-6-transglucosidase. Glycogen synthesis is promoted by insulin and glucocorticoids and by increased glucose concentrations. Glycogen synthetase is the rate-limiting step in glycogen synthesis and is converted from active to inactive form by phosphorylation by a cAMP-dependent protein kinase. Glycogenolysis , the breakdown of glycogen to release glucose units, is promoted by glucagon , epinephrine , vasopressin, angiotensin II, and oxytocin. Under conditions promoting glycogenolysis, phosphorylase catalyzes glycogenolysis by breakage of the linear 1-4 linkages. Phosphorylase is converted from the inactive to active form by phosphorylation by a cAMP-dependent protein kinase. Glucose molecules are removed from the 1-6 linkage branch points by the debrancher enzyme amylo-1-6-glucosidase (Fig. 11.2). Several inherited defects in various steps of glycogen metabolism have been identified, as shown in Fig. 11.4, and result in a clinical spectrum of glycogen storage diseases with the abnormal accumulation of glycogen in the liver .
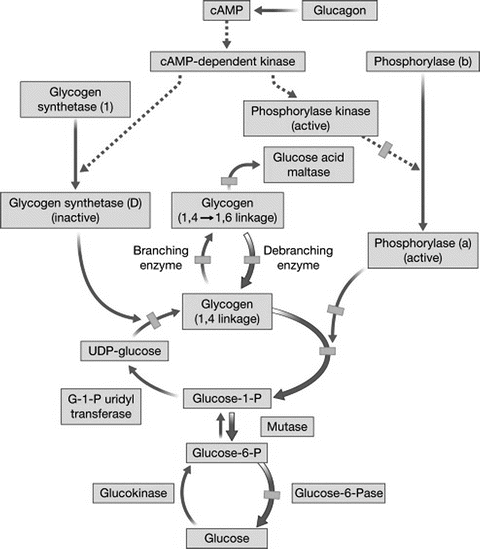
Fig. 11.4
Pathways for glycogen formation and glycogenolysis. Broken lines indicate enzymatic activation after glucagon stimulation; thick arrows indicate glycogen degradation from glucagon infusion, and hatched boxes indicate points in the metabolic sequence where enzymatic defects have been identified (Reproduced with permission from Elsevier [6])
4 Role of the Liver in Lipid Metabolism
Fatty acids are synthesized in the liver from carbohydrate precursors by conversion of these precursors to acetyl-CoA by the cytosolic fatty acid synthase complex. Hepatic fatty acid synthesis is stimulated by carbohydrate feeding and insulin. Fatty acids are generally stored in the liver as triglycerides, consisting of three fatty acids esterified to a glycerol backbone. Fasting, starvation, and diabetes mellitus with insulin deficiency cause increased hepatic fatty acid oxidation and production of acetoacetate and D-3-hydroxybutyrate (also known as ketones), which can be used as an energy source by muscle and brain. This process is known as ketogenesis . Lipolysis of adipose tissue triglycerides provides a major source of substrate fatty acids to the liver for production of ketones. The plasma glucagon to insulin ratio is probably the main regulator of ketogenesis.
The liver is a major site of fatty acid β-oxidation, which results in the production of energy using fatty acids as substrates. There are two postulated regulatory mechanisms for β-oxidation when carbohydrate is in short supply: regulation at the level of pyruvate formation and regulation at the level of fatty acid entry into the mitochondria, the site of most fatty acid oxidation, by way of carnitine acyltransferase I . Another site of fatty acid oxidation in the hepatocyte, particularly for saturated long chain fatty acids, is the peroxisome. Peroxisomal fatty acid oxidation is regulated only by fatty acid substrate concentration. This pathway may provide a mechanism for the production of acetyl CoA outside the mitochondria without the participation of citrate formed in the mitochondria. Peroxisomalβ-oxidation is inducible in rats by feeding a high-fat diet and treatment with certain hypolipidemic drugs that cause peroxisome proliferation.
The liver is a major site of cholesterol synthesis from acetyl CoA. In fact, all of the carbon atoms in cholesterol are derived from acetate. Cholesterol is ubiquitous throughout the body as a structural component of cell membranes and is a substrate for synthesis of several steroid hormones. Cholesterol is important in the liver as the precursor for bile acid synthesis. The rate-limiting step in cholesterol synthesis is the cleavage of CoA from the synthetic intermediate β-hydroxy-β-methylglutaryl (HMG) CoA with the simultaneous reduction to mevalonate . The enzyme which catalyzes this reaction is HMG-CoA reductase . Although the regulation of hepatic cholesterol synthesis is complex, several regulatory factors have been defined. In general, synthesis is up-regulated by any event that depletes the hepatocyte of cholesterol. For example, depletion of the bile acid pool by interruption of the enterohepatic circulation by biliary diversion, ileal resection or administration of a bile acid sequestrant such as cholestyramine will increase cholesterol synthesis, since bile acids are synthesized from cholesterol. Increased secretion of very low density lipoproteins (VLDL) stimulated by an influx of free fatty acids or carbohydrate requires cholesterol for packaging and depletes intracellular cholesterol resulting in increased synthesis. Hormones such as thyroid hormone, corticosteroids, and glucagon also up-regulate synthesis. Cholesterol synthesis is down-regulated by events that increase cellular cholesterol, such as the influx of cholesterol from receptor-mediated uptake of dietary cholesterol in chylomicron remnants and endogenous cholesterol in low density lipoproteins (LDL). The most potent down-regulators of cholesterol synthesis are oxidation products of cholesterol, 7-ketocholesterol and 25-hydroxycholesterol. Bile acid synthesis and biliary secretion of cholesterol are the major secretory pathways for cholesterol and will be subsequently discussed.
Triglycerides synthesized by the liver are secreted as particles called very low-density lipoproteins (VLDL). Lipoproteins are spherical particles, which serve as thermodynamically stable circulating packages for the transport of lipids through the aqueous environment of the bloodstream. The core of the particle contains hydrophobic, non-polar lipids, triglycerides and cholesteryl esters, and the surface coating consists of hydrophilic, polar lipids, phospholipid and free cholesterol , and lipid-binding peptides called apolipoproteins . Apolipoproteins generally serve important functions in the assembly, secretion, and peripheral metabolism of lipoprotein particles. The secreted hepatic VLDL serves to transport lipid from the liver to peripheral tissues. In humans, a large portion of the VLDL secreted is converted to low density lipoproteins (LDL), which are the major transporters of cholesterol to various tissues throughout the body. This pathway is summarized in Fig. 11.5.
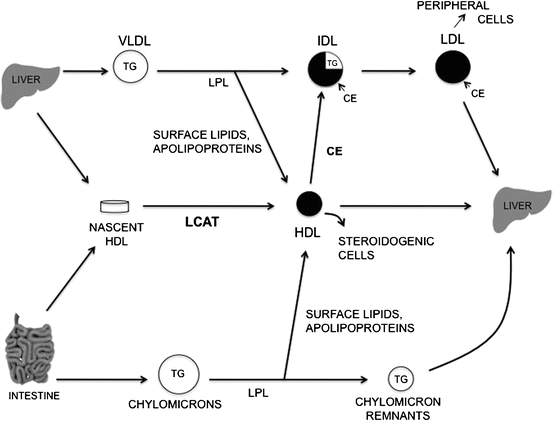
Fig. 11.5
Simplified scheme of plasma lipoprotein metabolism. Key events are the hydrolysis of triglyceride-rich lipoproteins by lipoprotein lipase (LPL), the conversion of VLDL to IDL, the movement of surface lipids and apolipoproteins released during lipolysis by LPL to HDL, the progressive enrichment of IDL and LDL with cholesteryl esters (CE) transferred from HDL as a result of the LCAT reaction, and the removal of LDL and chylomicron remnants by peripheral cells and the liver (Reproduced with permission from Glickman and Sabesin [7])
Carbohydrate feeding stimulates hepatic fatty acid production; thereby driving increased triglyceride and VLDL production and secretion. Secreted VLDL triglyceride is hydrolyzed by the enzyme lipoprotein lipase to liberate fatty acids for storage by adipose tissue and energy production by muscle. Lipoprotein lipase is bound to the endothelium of capillary beds in primarily adipose tissue and muscle and requires the apolipoprotein apo C-II, produced by the liver , as a cofactor. A related lipase produced by the liver, hepatic lipase, resides on sinusoidal endothelial cells. Hepatic lipase is important in HDL metabolism and in the hydrolysis of lipid in chylomicrons , VLDL and intermediate density lipoproteins (IDL).
The liver not only functions in the secretion of lipoprotein particles, but also is an essential organ for the uptake and metabolism of lipoproteins. Chylomicrons of intestinal origin, which carry mainly exogenous fatty acids and cholesterol, are metabolized by lipoprotein lipase to produce triglyceride-depleted, relatively cholesteryl ester-enriched remnants which are cleared by a receptor-mediated mechanism in the liver. This receptor-mediated clearance involves the LDL receptor and the LDL receptor-related protein, a membrane receptor, which binds and internalizes a wide variety of ligands. A significant proportion of plasma LDL particles are taken up by the liver via the LDL receptor, as the liver contains over half of the body’s total LDL receptors. Receptor-mediated LDL uptake results in an increase in esterification of the incoming cholesterol for storage, down-regulation of the number of LDL receptors, and suppression of cholesterol synthesis by the cell (Fig. 11.6). Finally, it has been demonstrated that the liver takes up a portion of VLDL particles shortly after they are secreted.
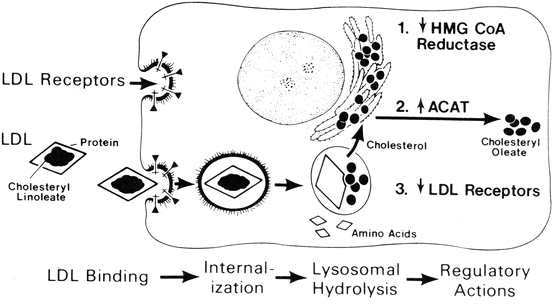
Fig. 11.6
Sequential steps in the LDL pathway. The numbers indicate the regulatory actions of LDL: 1 decrease in 3-hydroxy-3-methylbutyryl coenzyme A reductase (HMG CoA reductase) and cholesterol synthesis, 2 increase in acyl coenzyme A:cholesterol acyltransferase (ACAT) and cholesterol esterification, 3 decrease in the number of cell surface receptors for LDL (Reproduced with permission from Brown and Goldstein [4])
Recently, another key protein that regulates cellular cholesterol homeostasis in the liver through interaction with the LDL receptor was identified, proprotein convertase subtilisin kexin type 9 (PCSK9). Originally, PCSK9 was identified as a protein up-regulated during apoptosis of nerve cells. Subsequently, mutations of the PCSK9 gene associated with hypercholesterolemia were noted leading to identification of its role in cholesterol metabolism. Gain-of-function mutations result in increased plasma LDL cholesterol levels, and loss-of-function mutations cause the opposite. The normal function of PCSK9 appears to be interaction with the LDL receptor protein on the cell surface to interfere with normal recycling of the protein and direct it to a degradative pathway. This role of PCSK9 in cholesterol metabolism makes it an attractive therapeutic target for the treatment of hypercholesterolemia.
Another lipoprotein produced by the liver is high-density lipoprotein (HDL). This particle is secreted as a discoidal bilayer consisting primarily of phospholipid and free cholesterol . The major apolipoprotein is apo A-I, a cofactor for lecithin:cholesterol acyltransferase (LCAT), which is produced by the liver and catalyzes the transfer of a fatty acid from lecithin to free cholesterol to produce cholesteryl ester. These HDL particles acquire free cholesterol from peripheral tissues, which is then esterified and thereby moves into the core of the particle to change the shape from discoidal to spherical. This esterification is a driving force to effect net movement of cholesterol from tissue membranes to HDL. A portion of the cholesteryl ester in the HDL core may be transferred to a chylomicron , VLDL, or LDL particle through the action of a plasma cholesteryl ester transfer protein (CETP) to allow transport of the cholesterol to a variety of tissues throughout the body. The cholesterol from mature HDL particles is ultimately taken up by the liver via scavenger receptor-BI (SR-BI). Thus, HDL function in “reverse cholesterol transport ” to effect cholesterol movement from peripheral tissues to the liver. Serum levels of HDL cholesterol correlate inversely with risk for atherosclerotic coronary artery disease.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


