CHAPTER 89 Primary Biliary Cirrhosis
EPIDEMIOLOGY
PBC occurs worldwide and predominantly in women, with a female-to-male ratio of 9 : 1. The diagnosis of PBC usually is made between the ages of 30 and 60 years, with a range of 21 to 93 years. The disease has been documented in even younger patients—two teenagers 15 and 16 years of age, respectively.1 Until the early 1970s, PBC was considered a rare condition that manifested with persistent jaundice and almost inevitably progressed to end-stage liver disease. A better understanding of its pathogenesis, along with subsequent clinical and epidemiologic studies, has modified current concepts regarding this condition. PBC seems to be more common than was formerly believed because of increasing awareness of the disease and because asymptomatic patients are identified through the widespread use of screening tests such as determination of serum cholesterol levels and liver biochemical test levels in otherwise healthy persons.
The reported prevalence of PBC varies among countries, with a range of 19 cases per 1 million population in Israel to 402 cases per 1 million population in Olmsted County, Minnesota.2 Whether the difference in prevalence is real or a result of different methodologies used to detect the disease is unknown. Inconsistency in case definition and case finding methods, as well as imprecision in defining the study area, the populations evaluated, and the dates of diagnosis, particularly in earlier reports, makes comparisons among studies difficult. Estimates of the annual incidence of PBC range from 0.7 to 49 per 1 million population. Both the prevalence and incidence of PBC seem to have increased over time.2 The increase in prevalence possibly reflects an increase in survival time in patients with PBC.
In the United States,3 the age-adjusted reported incidence of PBC per 1 million person-years is 45 for women and 7 for men (27 overall). The reported prevalence per 1 million population is 654 for women and 121 for men (402 overall); these figures represent the highest prevalence rates for PBC ever reported.
PATHOGENESIS
Although the cause of PBC remains unknown, several lines of evidence suggest an autoimmune pathogenesis. The evidence includes the intense humoral and cellular response to an intracytoplasmic antigen, presence of highly specific antimitochondrial antibodies (AMA), involvement of T lymphocytes in the destruction of bile ducts, and numerous defects in immunologic regulation. Like other autoimmune diseases, PBC has a clear female predominance. Both PBC and the presence of AMA occur more frequently in close relatives of patients who have PBC than in controls. PBC is associated with an increased incidence of autoimmune disease of other types in both patients with PBC and their first-degree relatives.4,5 PBC seems to be triggered by an immune-mediated response to one or more allo- or autoantigens, which leads to progressive destruction of bile ducts, chronic cholestasis, and eventual biliary cirrhosis. Immunohistochemical phenotyping of inflammatory cells surrounding the bile ducts shows a combination of CD4+ and CD8+ T lymphocytes, accompanied by B lymphocytes and natural killer cells. Bile duct destruction is induced directly by the cytotoxicity of CD4+ and CD8+ T cells in contact with biliary epithelium. B lymphocytes are relatively uncommon in the inflammatory reaction but sometimes can be seen in clusters. Intracellular adhesion molecules (e.g., intracellular adhesion molecule-1 [ICAM-1]) are strongly expressed on many epithelial cells, particularly in areas of lymphocyte damage; these molecules may facilitate the interaction between destructive lymphocytes and their targets. In the early biliary lesions of PBC, eosinophilic infiltration and granulomas often are seen. PBC is principally a disease of the small intrahepatic bile ducts, with loss of biliary epithelial cells that line these ducts and resulting cholestatic damage. PBC is not restricted to the liver; abnormalities of salivary and lacrimal glands with an associated cellular phenotypic change similar to that seen in the biliary epithelial cells also occur. Three spontaneous autoimmune biliary disease mouse models6–8 and two induced models of PBC9,10 have been reported.
AUTOANTIBODIES
The mechanisms by which AMA directed to proteins located on the inner surface of mitochondrial membranes develop are unknown; PDC-E2 or a cross-reactive molecule is overexpressed on biliary epithelial cells in PBC, predominantly at the luminal domain, and PDC-E2–specific CD4+ T cells are present in portal inflammatory infiltrates of affected persons. Although AMA are predominantly of the IgG1 and IgG3 classes, most patients who have PBC exhibit polyclonal elevation of serum IgM levels; the IgM is not directed at mitochondrial or nuclear antigens. This phenomenon is suggestive of polyclonal activation of the B-cell compartment with an associated failure of isotype switching, representing aberrant B-cell activation.11 AMA do not appear to be cytotoxic: (1) They persist after liver transplantation without evidence of disease recurrence; (2) disease severity is unrelated to antibody titer; (3) they are not always present in PBC; and (4) they develop in animal models after the injection of recombinant PDC-E2 protein, but without resulting bile duct destruction or inflammation. Further, the different types and numbers of mitochondrial antigens recognized by Western immunoblot analysis at the time of the patient’s presentation is independent of the stage of the liver disease and not associated with specific clinical, biochemical, histologic, and immunologic features or with the Mayo risk score (see later).12
Antinuclear antibodies (ANA) are present in nearly one half of patients with PBC and in up to 85% of patients with AMA-negative PBC (see later). The most relevant immunofluorescent reactivities of ANA in patients with PBC are anti-multiple nuclear dots antibodies ([anti-MND], with the molecular target being a 100-kd soluble protein called Sp100), anticentromere antibodies, and antinuclear envelope antibodies. The immunofluorescence pattern of the antinuclear envelope antibodies is characterized as being rim-like and membranous; its molecular targets are structural components of the nuclear pore complex, such as gp210 and nucleoprotein p62, and of the nuclear membrane, such as lamin B receptors. Antibodies against the nuclear pore protein gp210 (anti-gp210) are found in 25% of patients with AMA-positive PBC and in up to 50% of those with AMA-negative PBC. ANA with the MND and rim-like and membranous patterns, which are relatively rare or absent in normal and pathologic controls, are strongly associated with PBC and can be considered to be surrogate markers of PBC in AMA-negative patients.13–15 The specificity of anti-gp210 for PBC when detected by immunoblotting is greater than 99%, whereas antibodies to p62 (anti-p62) are found in approximately 25% of patients and are highly specific for PBC. Anti-p62 antibodies seem to be mutually exclusive with anti-gp210 antibodies. Further, anti-gp210 and possibly anti-p62 also offer prognostic information in that they seem to be associated with aggressive disease with a poor prognosis.13,16,17 In Japanese patients with PBC, anticentromere antibodies are associated with the development of portal hypertension.18
GENETIC FACTORS
The occurrence of PBC in relatives of affected persons plus abnormalities of cell-mediated immunity in first-degree relatives of patients with PBC suggests a genetic association. This association is further supported by the finding that PBC exhibits a higher concordance rate in monozygotic than dizygotic twin pairs, suggesting a genetic component to disease susceptibility and expression of the susceptibility through genes that regulate the immune response.19 Many of the familial risk and genetic studies of PBC have failed to distinguish, however, between true genetic risk and shared environmental exposure.
Although no link between PBC and a specific human leukocyte antigen (HLA) class I phenotype has been found, HLA class II molecules may contribute to the development of this condition. The HLA associations with PBC detected most commonly have been with the DRB1*0801 allele in European and North American white populations and DRB1*0803 in Japanese populations.20,21 Although the DRB1*08 allele seems to impart a significant risk for PBC, with odds ratios of 3 or higher in white and Japanese populations, a study from China reported that the frequencies of the DRB1*0701 and DRB1*03 alleles were increased significantly in patients with PBC compared with controls, with no difference in the frequencies of the DRB1*08 allele.22 A few class II HLA alleles demonstrate protective associations against PBC. These alleles include DQA1*0102 in United States and Japanese studies and DQB1*0602 in a United States study. More recently, DRB*13 was found to be protective against PBC in patients from the United Kingdom and Italy, and DRB1*11 was protective in patients from Italy but not those from the United Kingdom.20,23 An association with HLA class III genes, which code for complement components C2 and C4, cytokines, and complement factor B, has been studied less extensively. Earlier studies reported an increased frequency of haplotype C4B2 and haplotype C4A*Q0 in patients with PBC. Smaller, older studies of the association between PBC and alleles affecting the expression of tumor necrosis factor-α have been reported, but an association remains unresolved.
MOLECULAR MIMICRY
Molecular mimicry between host autoantigens and unrelated exogenous proteins is one of the hypotheses to explain how autoantibodies to self-proteins arise, break tolerance, and lead to autoimmune disease. Molecular mimicry of an extrinsic protein produced by an infectious agent has long been suggested as a possible initiating event in PBC. Infectious agents incriminated in the immune response in PBC include various bacteria and viruses, most recently Chlamydia pneumoniae,24 Novospingobium aromaticivorans,25 and human betaretrovirus.26 Microorganisms produce a multitude of foreign antigens that collectively constitute the major determinants recognized by the immune system. These antigens potentially include a variety of carbohydrates, lipids, and proteins that can be recognized by specific receptors on inflammatory cells. In PBC, PDC-E2 appears to be an ideal candidate for foreign antigens to mimic. PDC-E2, particularly its inner lipoyl domain, is highly conserved among bacteria, yeasts, and mammals. Autoimmune phenomena in PBC could result from peptides that mimic T-cell epitopes of microbial proteins and that are derived from, and presented by, abnormally expressed HLA class II molecules. Molecular mimicry has been invoked to explain the breaking of tolerance against mitochondrial antigens. Definitive evidence for this theory is still lacking, however.
XENOBIOTICS AND OTHER IMPLICATED AGENTS
Xenobiotics are foreign compounds that may alter self-proteins by inducing a change in the molecular structure of the native protein sufficient to induce an immune response. The immune response may then result in the recognition of both the modified and the native proteins.27 The continued presence of the self-protein may perpetuate the immune response initiated by the xenobiotic-induced adduct, thereby leading to chronic autoimmunity. Because many xenobiotics are metabolized in the liver, the potential for liver-specific alteration of proteins is substantial. To address the hypothesis that PBC is induced by xenobiotic exposure, Long and colleagues28 synthesized the inner lipoylated domain of PDC-E2, replaced the lipoic acid moiety with synthetic structures, and quantified the reactivity of these structures with sera from patients with PBC. AMA from all patients reacted more strongly to 3 of the 18 modified organic autoepitopes than to the native domain. Defective sulfoxidation of certain compounds, such as bile acids, estrogen, or drugs, and selenium deficiency have been proposed as underlying mechanisms that may lead to this process. These hypotheses remain unproved.
CLINICAL FEATURES
SYMPTOMATIC DISEASE
The typical patient with symptomatic disease (Table 89-1) is a middle-aged woman with a complaint of fatigue or pruritus. Other symptoms include right upper quadrant abdominal pain, anorexia, and jaundice. Fatigue, although relatively nonspecific, is considered to be the most disabling symptom by many patients, and it worsens in some patients as the disease progresses.29,30 Fatigue in patients with PBC does not correlate with several markers of disease severity, or with the patient’s age or thyroid status, but does correlate with sleep disturbance and depression.29 In a four-year follow-up study, the severity of fatigue was quite stable overall; only liver transplant recipients experienced a significant improvement in fatigue.31 In that study, fatigue was also found on multivariate analysis to be an independent predictor of mortality, particularly cardiac death.31
Table 89-1 Symptoms and Signs of Primary Biliary Cirrhosis at Presentation
| SYMPTOM OR SIGN | FREQUENCY (%) |
|---|---|
| Fatigue | 21-85 |
| Pruritus | 19-55 |
| Hyperpigmentation | 25 |
| Hepatomegaly | 25 |
| Splenomegaly | 15 |
| Xanthelasma | 10 |
| Jaundice | 3-10 |
| Right upper quadrant pain | 8 |
| None | 25-61 |
Pruritus may occur at any point, early or late, in the course of the disease, or intermittently throughout the course. Pruritus generally is intermittent during the day and is most troublesome in the evening and at night. Pruritus often resolves as the disease progresses, but in some patients, severe, intractable pruritus can develop in earlier stages of the disease and may require liver transplantation for effective management. In a population-based study of 770 patients with PBC from England, the cumulative risk of developing pruritus was 19%, 45%, and 57% at 1, 5, and 10 years, respectively.30
Most patients with PBC do not have jaundice at the time of diagnosis. Jaundice occurs later in the course of the disease and usually is persistent and associated with a worse prognosis. Symptoms also may relate to fat-soluble vitamin deficiency, bone pain with or without spontaneous fractures, or an associated autoimmune disease that may occur in patients with PBC (Table 89-2). Symptoms and signs of advanced liver disease, such as ascites, bleeding from gastroesophageal varices, and encephalopathy, usually occur late in the course of PBC.
Table 89-2 Diseases Associated with Primary Biliary Cirrhosis
| DISEASE | FREQUENCY (%) |
|---|---|
| Keratoconjunctivitis sicca (Sjögren’s syndrome) | 72-100 |
| Renal tubular acidosis | 50-60 |
| Arthritis/arthropathy | 4-42 |
| Gallstones | 33 |
| Autoimmune thyroiditis | 15-20 |
| Scleroderma and its variants | 15-19 |
| Raynaud’s disease | 8 |
| CREST or any of its components | 7 |
| Scleroderma | 3-4 |
| Cutaneous disorders—lichen planus, discoid lupus, pemphigoid | 11 |
| Hepatocellular carcinoma | 1-2 |
| Pulmonary fibrosis | Rare |
| Celiac disease | Rare |
CREST, calcinosis, Raynaud’s phenomenon, esophageal dysmotility, sclerodactyly, telangiectasia.
ASSOCIATED DISEASES
Many of the diseases found frequently in patients with PBC (see Table 89-2) are thought to be related to disturbances in immune mechanisms. These associated disorders include Sjögren’s syndrome (characterized by dry eyes [keratoconjunctivitis sicca] and dry mouth), scleroderma and its variants, rheumatoid arthritis, some cutaneous disorders, renal tubular acidosis, and thyroiditis.
DIAGNOSIS
The diagnosis of PBC is established by liver biochemical test results consistent with chronic cholestasis plus the presence in serum of AMA. Liver biopsy helps to confirm the diagnosis of PBC but may not be necessary for establishing the diagnosis in patients with characteristic chronic cholestasis and AMA.31
BIOCHEMICAL FEATURES
Liver biochemical test results show a cholestatic picture. Almost all patients have increased serum levels of alkaline phosphatase (three to four times the upper limit of normal) and gamma glutamyl transpeptidase. Serum aminotransferase (aspartate aminotransferase [AST], alanine aminotransferase [ALT]) levels are mildly elevated (usually less than three times normal); marked elevations (more than five times normal) are distinctly unusual and may suggest PBC-autoimmune hepatitis overlap syndrome (see Chapter 88) or coexisting viral hepatitis. Serum bilirubin levels usually are normal in early stages and increase slowly over the course of the disease; levels ultimately may exceed 20 mg/dL. A high serum bilirubin level, low serum albumin, and prolonged prothrombin time indicate a poor prognosis and advanced disease. Serum immunoglobulin levels, especially IgM, are increased, as are serum levels of bile acids, in particular cholic and chenodeoxycholic acids, and cholesterol.
HISTOPATHOLOGIC FEATURES
The initial lesion on a liver biopsy specimen in PBC (Figs. 89-1 and 89-2A and B) is damage to epithelial cells of the small bile ducts. The most important and only diagnostic clue in many cases is ductopenia, defined as the absence of interlobular bile ducts in greater than 50% of portal tracts. The florid duct lesion, in which the epithelium of the interlobular and segmental bile ducts degenerates segmentally, with formation of poorly defined, noncaseating epithelioid granulomas, is nearly diagnostic of PBC but is found in a relatively small number of cases, mainly in early stages.
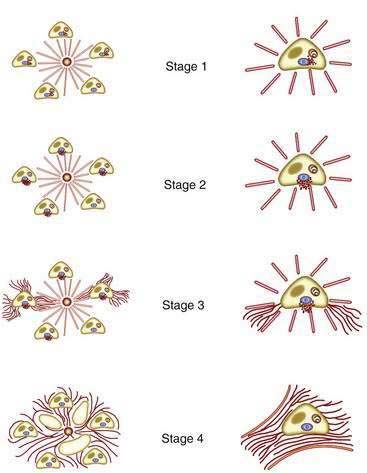
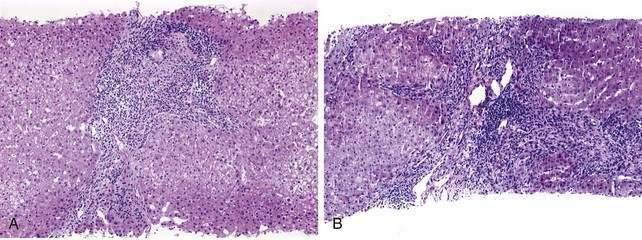
The two most popular histologic staging systems are those proposed by Ludwig and colleagues and Scheuer, which classify the disease in four stages. Both systems describe progressive pathologic changes, beginning initially in the portal areas surrounding the bile ducts and culminating in cirrhosis. Ludwig stage 1 disease is characterized by inflammatory destruction of the intrahepatic septal and interlobular bile ducts that range up to 100 µm in diameter. These lesions often are focal and described as florid duct lesions, characterized by marked inflammation and necrosis around a bile duct. The portal tracts usually are expanded by lymphocytes, with only sparse neutrophils or eosinophils seen. In stage 2 disease (see Fig. 89-2A), the inflammation extends from the portal tract into the hepatic parenchyma, a lesion called interface hepatitis, or formerly, piecemeal necrosis. Destruction of bile ducts with proliferation of bile ductules can be seen. Stage 3 disease is characterized by scarring and fibrosis. Lymphocytic involvement of the portal and periportal areas, as well as the hepatic parenchyma, can be seen, but the hallmark of this stage is the presence of fibrosis without regenerative (or regenerating) nodules. Stage 4 disease is characterized by cirrhosis with fibrous septa and regenerative nodules (see Fig. 89-2B).
Most patients with PBC demonstrate progression of the liver disease; a few patients have a prolonged course of histologic stability, and only rare patients have sustained regression. A time course Markov model has been used to describe the rate of histologic progression over time (Table 89-3).







Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


