Adenomatous polyposis syndromes
Familial adenomatous polyposis (FAP)
MYH-associated polyposis (MAP)
Turcots syndrome
Lynch syndrome
Hamartomatous polyps
Solitary juvenile polyp
Juvenile polyposis syndrome
PTEN hamartoma tumor syndrome
Peutz–Jeghers syndrome
Inflammatory polyps
Clinical Presentation of Gastrointestinal Polyps
The most common clinical manifestation of a large bowel polyp is painless rectal bleeding . Other symptoms attributed to polyps include abdominal pain, altered bowel habit, and prolapse of polyp or rectum. Diagnosis will be made upon full colonoscopy with polypectomy. The clinical presentation, endoscopic appearance, pathological findings, histological description of the polyp, and genetic investigations are all necessary to establish the correct diagnosis of a polyposis syndrome. Once a polyp has been identified at endoscopy, a carefully targeted family history must be taken to enquire if there are family members who have or have had cancer, the site of the cancers, and age of onset (see Table 52.2). Taking such a history to develop a detailed family cancer pedigree may require the expertise of a multidisciplinary familial cancer clinic or polyposis registry [1] .
Table 52.2
History and examination in a child with possible GI polyps
History |
Nature of bleeding and frequency |
Painful or painless rectal bleeding |
History of GI obstructive symptoms |
Detailed family history exploring early deaths or diagnosis of GI cancer and include history of non-GI malignancies |
Weight loss, anorexia (tumor) |
Learning difficulties (JPS or PTEN hamartoma) |
Examination |
Mucosal pigmentation (PJS) |
Dysmorphic features (JPS) |
Edema (hypoalbuminemia in infantile JPS) |
Extra intestinal manifestations of FAP |
Hepatic mass (FAP) |
Cutaneous telangiectasia (HHT with JPS) |
Thyroid mass (FAP or Cowden) |
JPS juvenile polyposis syndrome, PTEN phosphatase and tensin, PJS Peutz–Jeghers syndrome, FAP familial adenomatous polyposis, HHT hereditary hemorrhagic telangiectasiaThe Single Hamartomatous Polyp (the Juvenile Polyp)
With a prevalence of up to 3 % of children, the commonest presentation of a single hamartomatous (or juvenile) polyp is painless bright red rectal bleeding with blood seen on wiping or mixed in the stool. Seventy percent of juvenile polyps are found in the rectum or rectosigmoid. The rest however are found more proximally, hence the need for pancolonoscopy. Single or solitary juvenile polyps are benign and confer no future risk of malignancy. Presently, it is unknown whether children who present with a single polyp in childhood may continue to form polyps over time [2].
If a polyp is found to be solitary after full colonoscopy, and there is no relevant family history, endoscopic polypectomy should be sufficient treatment. After polypectomy, parents should be advised that juvenile polyps may be the first feature of a hamartomatous polyposis syndrome, and if new symptoms arise, the child should be reinvestigated. If multiple juvenile polyps are found (> 3–5) or there is a positive family history (e.g., colonic polyps or early onset CRC), a hamartomatous polyposis syndrome should be considered and an alternative approach should be taken .
Hamartomatous Polyposis Syndromes
Hamartomatous polyposis syndromes are a rare group of hereditary autosomal dominant disorders. The polyps themselves are benign but the polyposis syndromes confer significant potential for developing CRC and extra colonic cancers. The hamartomatous polyposis syndromes include juvenile polyposis syndrome (JPS), Peutz–Jeghers syndrome (PJS) , and phosphatase and tensin (PTEN) hamartoma tumor syndrome, which includes Cowden (CS) and Bannayan–Riley–Ruvalcaba syndrome (BRRS).
Juvenile Polyposis Syndrome
JPS is rare, with an estimated prevalence of 1 in 100,000 individuals presenting with multiple hamartomatous polyps and an increased risk of gastrointestinal malignancies. Affected individuals develop multiple gastrointestinal polyps which are predominantly in the colon, though other areas of the gastrointestinal tract can also be involved. The condition should be considered in any patient with more than five juvenile polyps in the colon, any juvenile polyps found in other parts of the gastrointestinal tract, or if any juvenile polyp is found in a child with a positive family history. Patients present with chronic and acute gastrointestinal bleeding, anemia, prolapsed rectal polyps, abdominal pain and diarrhea and can develop up to 50–200 polyps in the colon, while in some patients, with generalized juvenile polyposis, polyps can affect the colon, stomach, and small bowel. The total number of polyps needed to make the diagnosis remains controversial, between 3 and 5 [3].A significant proportion of patients with juvenile polyposis have been reported to have other morphological abnormalities including digital clubbing, polydactyly, macrocephaly, alopecia, cleft lip or palate, congenital heart disease, and mental retardation. A specific variant of JPS called juvenile polyposis of infancy has an onset in infancy presenting with anemia and hemorrhage, diarrhea, protein-losing enteropathy, intussusception , and rectal bleeding. The course in such infants is fulminant and, even despite colectomy, in severe cases, death occurs before the age of 2 years.
There is little doubt that juvenile polyposis is a premalignant condition. There is a 15 % incidence of colorectal carcinoma occurring in patients under the age of 35 years, leading to a cumulative lifetime risk of CRC of 38–68 % with a mean age of colonic neoplasia onset between 38 and 44 years [4].
Genetics of Juvenile Polyposis
JPS is a fully penetrant condition with variable expression. Sixty percent of cases are familial and the others occur sporadically. Germline mutations in SMAD4, BMPR1A, and ENG1 cause JPS. Approximately 54 % of JPS cases will have a detectable mutation [5] .
Patients with the SMAD4 mutation on chromosome 18q21.2 appear to have a higher risk of gastric polyps and hereditary hemorrhagic telangiectasia (HHT). The latter condition is characterized by cutaneous telangiectasia and risk of arteriovenous malformations . Patients found to have the SMAD4 mutation should be screened for cerebral and pulmonary arteriovenous malformations associated with HHT [6]. BMPR1A is located on chromosome 10q22.3 and accounts for another 20 % of JPS patients. ENG1 mutation on gene 9q34.1 has recently been described in JPS patients without HHT.
Screening and Follow-Up
Once the gene mutation has been identified in the index patient, other at risk family members should be tested. All children of an affected adult will have a 50 % chance of inheriting the mutation, and if the family mutation is known, children should undergo genetic screening.
Proposed guidelines suggest affected children should undergo colonoscopic surveillance every 2 years or earlier if symptoms arise [7]. In those families where the gene mutation is not known, first-degree relatives of patients with JPS should be screened by a single colonoscopy starting at age 12–15 years, even when the subject is asymptomatic (Fig. 52.1).
Fig. 52.1
Screening and surveillance algorithm for juvenile polyposis syndrome
Full colonoscopy is necessary as right-sided polyps are the most common. All polyps should be resected. Annual colonoscopy is performed until all polyps have been resected after which the screening interval is stretched to every 2–3 years. Gastroscopy is commenced from mid-teens. Colectomy is warranted for patients with cancer, dysplasia, or high polyp burden with symptoms that cannot be controlled endoscopically.
PTEN Hamartoma Tumor Syndrome
This group comprises three rare genetic syndromes: Cowden syndrome, BRRS syndrome, and Proteus syndrome (the latter has very few intestinal features). All are associated with a mutation in the PTEN gene located at 10q23.3 [8]. All three syndromes are characterized more by extraintestinal manifestations than intestinal polyposis. The PTEN mutation can be detected in 80 % of patients with Cowden, 60 % with BRRS, and 50 % with Proteus syndrome.
Cowden syndrome rarely presents in childhood. Clinical manifestations include macrocephaly, papillomatous papules, mucocutaneous lesions, facial trichilemmoma, and acral keratosis. It carries a 50 % risk of breast cancer in adult women and a 10 % lifetime risk of epithelial thyroid cancer. Guidelines presently recommend screening for breast, thyroid, endometrial, and kidney cancer from the age of 18–25 years.
BRRS presents in childhood with gastrointestinal hamartomas, particularly in the ileum and colon, which cause intussusception, rectal bleeding, and hypoalbuminemia . There are additional characteristics including macrocephaly, developmental delay, abnormal metacarpal and phalanges, pectus excavatum, scoliosis, genital pigmentation, and hemangiomatosis. BRRS presents prior to adolescence , and there is value in genetic testing in early childhood in a family where the mutation has been identified. Patients with BRRS need regular colonoscopy and small-bowel surveillance as they are at risk of anemia, intussusception, and hypoalbuminemia from the polyposis. They carry a probable lifetime increased risk of cancer, and surveillance is recommended from age 18–25 years focusing on renal, thyroid, and breast cancer [9].
Peutz–Jeghers Syndrome
Clinical Features and Diagnosis
PJS is a rare autosomal dominant condition with a prevalence of 1 in 50,000 to 1 in 200,000 live births . It is characterized by mucocutaneous pigmentation and the presence of hamartomatous polyps throughout the gastrointestinal tract. Polyps arise primarily in the small bowel and to a lesser extent in the stomach and colon. Polyps are most commonly found in the jejunum and cause bleeding and anemia, or intussusception and obstruction from an early age. Presumptive diagnosis can be made in those with a positive family history and typical PJS freckling.
Pigmentation tends to arise in infancy, occurring around the mouth, nostrils, perianal area, fingers, toes, and the dorsal and volar aspects of hands and feet. The primary concern to the pediatrician is the risk of small-bowel intussusception causing intestinal obstruction, vomiting, and pain (Fig. 52.2). In addition, intestinal bleeding—with melena, hematemesis, and rectal bleeding—can occur, leading to anemia .
Fig. 52.2
Large obstructing duodenal PJS polyp seen at gastroscopy
Genetics of PJS
As with other hamartomatous syndromes , PJS has an autosomal dominant pattern of inheritance, and many cases may be sporadic new mutations. The mutated gene STK11(LKB1), located on chromosome 19p 13.3, can be identified in up to 90 % of PJS patients [10]. After appropriate genetic counseling and informed consent, testing at-risk family members may be performed early in childhood so that gastrointestinal surveillance can commence before gastrointestinal complications arise.
Screening, Management, and Complications
Individuals at risk of PJS should be evaluated in infancy for pigmented lesions and gastrointestinal symptoms. Asymptomatic at risk children should undergo genetic testing for the family proband mutation in the STK11/LKB1 mutation if known, soon after infancy, so the family can access medical care early if the child develops symptoms consistent with small-bowel obstruction.
The management of a young child with mid-gut PJS polyps is controversial, but recommended guidelines have been published [11]. Children who present with mid-gut complications need polypectomy either by laparotomy and intraoperative enteroscopy (IOE; Fig. 52.3) or double balloon enteroscopy (DBE). An IOE is recommended in any patient with PJS undergoing laparotomy, as careful endoscopy via an enterotomy in the small bowel allows identification and removal of polyps, thus avoiding multiple enterotomies and the risk of short bowel syndrome associated with resection. This technique is superior to palpation and transillumination in identifying polyps, and removal of all detected polyps (“clean sweep”) reduces re-laparotomy rate significantly [12].
Fig. 52.3
Intraoperative enteroscopy performed at laparotomy for intussusceptions in a patient with PJS
DBE with polypectomy of PJS polyps in the small bowel carries a significant risk of perforation and should be performed only by those who are expert in polypectomy. Muscularis mucosa commonly invaginates into the large pedunculated stalk increasing the risk of perforation at electrocautery (Fig. 52.4). DBE can be combined with laparoscopy to assess perforations that may arise at polypectomy.
Fig. 52.4
Indentation of the serosal surface of the ileum from intusscepting PJS polyp
Endoscopic evaluation of the upper and lower gastrointestinal tract and imaging of the small bowel should be performed from the age of 8 years. Screening should start earlier if symptoms are present before this. The development of video capsule endoscopy (VCE) has replaced barium enterography as the preferred technique for assessing the small bowel [13]. VCE is more sensitive, preferred by patients, and reduces the lifetime risk from cumulative radiation exposure. An acceptable alternative to VCE is MRI enterography with a close correlation between the two modalities, especially with polyps > 15 mm [14]. The advantages and disadvantages of prophylactic polypectomy for asymptomatic patients should be discussed with the family (Fig. 52.5). Prophylactic polypectomy of larger small-bowel polyps (> 1.5 cm) by intraoperative or DBE should be performed in order to reduce the incidence of subsequent complications and the requirement for emergency laparotomy.
Fig. 52.5
Screening and surveillance algorithm for Peutz–Jeghers syndrome MRE magnetic resonance enterography, DBE double-balloon enteroscopy, IOE intraoperative enteroscopy
The risk of neoplasia is well documented in young adults. A meta-analysis to assess the risk of cancer in PJS identified a relative risk for all cancers in PJS patients (aged 15–64) of 15.2 compared to the normal population, with tumors. Clinicians caring for adolescents with PJS should be aware of unusual symptoms and have a low threshold for investigating potential malignancies. A recommended screening program for PJS patients after adolescence is shown in Table 52.3.
Table 52.3
Suggested program for screening for malignancies in Peutz–Jeghers syndrome after adolescence
General |
Annual hemoglobin and liver function tests |
Annual clinical examination |
Genital tract |
Annual examination and testicular US biennial from birth until 12 years |
Cervical smear every 3 years from age 25 years |
Gastrointestinal |
Baseline EGD/colonoscopy age 8 |
Polyps detected, continue 3 yearly until 50 years |
No polyps detected, repeat age 18 years, then every 3 years until 50 years |
VCE every 3 years from age 8 years |
Breast |
Monthly self examination from age 18 years |
Annual breast MRI from age 25–50, thereafter annual mammography |
EGD esophagogastroduodenoscopy, VCE video capsule endoscopy, MRI magnetic resonance imaging Adenomatous Polyposis Syndromes
Familial Adenomatous Polyposis
In children, GI adenomas are almost always associated with hereditary adenomatous polyposis syndromes . FAP is characterized by the development of hundreds of adenomas in the colon and rectum as well as several extra colonic manifestations. Almost all affected patients will develop aCRC if not detected and treated at an early stage.
Clinical Features
Patients with FAP typically develop multiple adenomas throughout the large bowel—usually more than 100 and sometimes more than 1000 (Fig. 52.6) . Polyps begin to appear in childhood or adolescence and increase in number with age. The standard clinical diagnosis of typical/classical FAP is based on the identification of > 100 colorectal adenomatous polyps. By the fifth decade, CRC is almost inevitable if colectomy is not performed. Attenuated FAP (AFAP) is a milder form of the disease which is observed in 8 % of cases. It is characterized by fewer adenomas and later presentation [15].
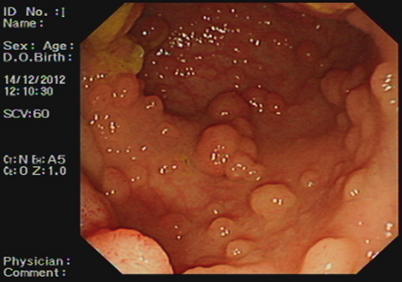
Fig. 52.6
Endoscopic appearance of large adenomatous polyps seen in advanced FAP
Gastric fundic gland polyps occur in the antrum in 50 % of FAP-affected adults, and small-bowel adenomas also occur and neither requires intervention. Children under 5 years of age may develop hepatoblastoma , with an increased risk in boys [16]. Adult patients are at increased risk of malignancies of the duodenum and ampulla of Vater, with a lifetime risk of duodenal adenomas of 100 %. Duodenal adenomas will progress to malignancy if untreated in 5 % of adults and are the second most common malignancy in FAP and AFAP . In addition, FAP is associated with an increased risk of cancers of the thyroid, brain, and pancreas while papillary carcinoma of the thyroid has been reported in adolescence.
Extraintestinal manifestations are common (see Table 52.4). Pigmented ocular lesions (previously termed congenital hypertrophy of the retinal epithelium or CHRPE) are found in some but not all cases .
Table 52.4
Extracolonic manifestations of FAP in children and young adults
Site | Examples |
|---|---|
Bone | Osteomas, mandibular, and maxillary |
Exostosis | |
Sclerosis | |
Dental abnormalities | Impacted or supernumerary teeth |
Unerupted teeth | |
Connective tissue | Desmoid tumors |
Excessive intra-abdominal adhesions | |
Fibroma | |
Subcutaneous cysts | |
Eyes | Congenital hypertrophy of the retinal pigment epithelium |
CNS | Glioblastomas, for example, Turcot’s syndrome |
Adenomas | Stomach |
Duodenum | |
Small intestine | |
Adrenal cortex | |
Thyroid gland | |
Carcinomas | Thyroid gland |
Adrenal gland | |
Liver | Hepatoblastoma |
Genetics of FAP
FAP is an autosomal dominant inherited condition caused by a mutation in the adenomatous polyposis coli (APC) gene occurring in 1:10,000 births. In 20–30 % of cases, the condition is caused by a spontaneous mutation with no clinical or genetic evidence of FAP in the parents or family .
The gene responsible for FAP, APC, is located on chromosome 5q21 and appears to be a tumor-suppressor gene [17]. Most mutations are small deletions or insertions which result in the production of a truncated APC protein which then predisposes to adenoma formation. Many mutations have been identified on this large gene, and there is a correlation between the genetic site and severity of clinical manifestation. Mutations between codons 1250 and 1464 (Fig. 52.7), and especially those with a mutation at codon 1309, are associated with a severe form of FAP. Mutations localized at the extreme ends of the gene and in the alternatively spliced part of exon 9 are associated with a mild form of FAP, and an intermediate expression of disease is found in patients with mutations in the remaining parts of the gene. Other phenotype–genotype correlations have been observed [18]. These correlations are not absolute, and there may be considerable intrafamilial variation suggesting that there are other factors involved in the pathogenesis of the disease. Families need to be aware that the mutation may only be detected in 70–90 % of cases .
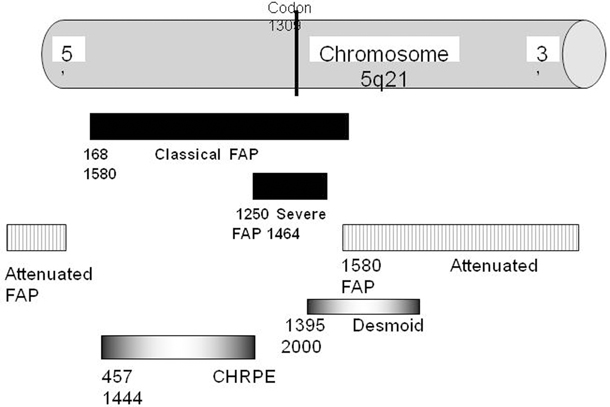
Fig. 52.7
APC protein domains showing FAP genotype-phenotype correlation with codon number APC adenomatous polyposis coli, FAP familial adenomatous polyposis, CHRPE congenital hypertrophy of the retinal epithelium
Diagnosis: Interpretation of the Genetic Test and Clinical Screening in FAP
In order to determine the appropriate screening protocol for a given family, the first step would be to seek which mutation is present in the FAP affected index case. If a mutation cannot be found, the genetic testing is non-informative, and it will not be possible to offer predictive testing to asymptomatic at-risk relatives .
When family-specific mutation is identified, directed DNA diagnostic techniques can be readily used to predict FAP in other family members. The absence of the gene mutation in other family members is considered accurate in excluding FAP, and the subject should be considered to hold an average population risk for the subsequent development of adenomas and cancer. Such genotype negative individuals can be discharged from follow-up.
The presence of the family-specific gene mutation confirms the diagnosis of FAP, and such patients should undergo endoscopic assessment. The diagnosis is confirmed by finding polyps upon sigmoidoscopy or colonoscopy which are histologically confirmed as adenomas (see Fig. 52.2). Teenagers predicted at gene testing to be affected will require a full colonoscopy by the age of 14–16, preferably beginning at age 10–12 years [19], to determine polyp density and location, and degree of dysplasia. Upper endoscopic surveillance of the stomach, duodenum, and periampullary region with a side-viewing endoscope is recommended after the age 20 years, unless the patient has symptoms such as upper abdominal pain which warrant earlier investigation .
For families in which the genotype is not known, protocols vary. It is necessary to perform annual sigmoidoscopy on all first-degree relatives until adenomas are found .
No patient should undergo screening for FAP without detailed counseling. The individual being screened must understand the nature of the test and its possible outcomes. Many authorities feel that the child should be involved in the decision-making process, and the diagnosis be delayed until the child is old enough to contribute to the screening program, for example, from the age of 11 years onwards [20]. Although parents might request testing of a child or infant at a young age, there is value in deferring a test until the child or adolescent can participate in the discussion [21]. Well-informed consent, as a mature minor, prior to predictive testing is the most desirable outcome. There are psychological issues, as well as family, insurance, and employment implications, which may arise in the case of a positive result. These should be discussed prior to testing, and there should be a clear protocol for posttest management [22].
The current advice is to commence endoscopic surveillance from the early teens. Some patients, especially those with a mutation located at codon 1309 in the APC gene, may develop severe polyposis of the colorectum before the age of 10, and therefore, attention must be paid to FAP-related symptoms. These symptoms may include increasing bowel movements, looser stools, mucous discharge, rectal bleeding, abdominal, or back pain. In symptomatic patients, endoscopic investigation may be indicated at any age. Severe dysplasia and even malignancy have been documented in children with FAP under the age of 12 years. Consequently, those children from families in which severe dysplasia or carcinomas have been found at a young age should undergo screening at an earlier age [23]. Some clinicians advocate annual screening for hepatoblastoma with liver palpation, ultrasound, and serum á-fetoprotein levels in at-risk children between the ages of 0–6 years .
Management of FAP
Internationally agreed guidelines are currently in place for the management of patients with FAP [24]. If adenomas are detected, colonoscopic investigations should be performed annually until colectomy is planned [25]. Colectomy is currently the only effective therapy that eliminates the inevitable risk ofCRC . In the absence of severe dysplasia, colectomy is usually performed in mid- to late teens or early 1920s to accommodate work and school schedules. Almost all screen-detected adolescents are asymptomatic and therefore may not be willing to contemplate interruptions in their schooling or effects on relationships. The surgical option, therefore, must not only be carefully timed but also have low morbidity and excellent functional result .
Colectomy is indicated as soon as there are more that 100 adenomas (measuring up to 5 mm) or adenomas showing a high degree of dysplasia. The timing of primary preventative surgery may be influenced by knowledge of the mutation site and the likely severity of the polyposis. For example, patients with a deletion at codon 1309 should be offered earlier surgery since this phenotype is characterized by a large numbers of polyps and a higher risk of cancer [26] .
Surgical options are either subtotal colectomy with ileorectal anastomosis (IRA), or restorative proctocolectomy with ileal pouch anal anastomosis (IPAA or pouch procedure). The IRA is a low risk operation with good functional results and can be performed laparoscopically. There is no pelvic dissection, and therefore, attendant risks of hemorrhage, loss of fertility in women, and damage to adjacent organs such as the ureters are avoided. Complication rates after IRA are low, and postoperative bowel function is almost always good, averaging four semi-formed stools daily. After the IRA, the rectum remains at risk of cancer. Therefore, postoperative six-monthly surveillance of the rectum is mandatory.
An IPAA procedure removes the CRC risk almost completely, but is more complicated than an IRA. It carries a higher morbidity and risk of complications. It often requires a temporary loop ileostomy, reoperations and longer hospital stays, night evacuation and decreased fertility in women [27]. Bowel frequency is generally higher than that for an IRA. The risk of cancer does exist after IPAA as they may develop at the anastomosis or below. The pouch should be examined regularly postoperatively for adenomas [28] .
The advantages of an IPAA with a lower risk of cancer must be balanced against the higher operative morbidity. Conversion after an IRA to an ileoanal pouch can be carried out when the patient is older [29]. An IPAA is the treatment of choice if the patient has a large number of rectal adenomas, for example, > 15–20 adenomas, if there is presence of adenoma with severe dysplasia, a colon with > 1000 adenomas or those with high-risk genotypes (e.g., codon 1309) [30, 31]. In patients with only a few rectal adenomas or with a polyp-free rectum, both options are possible although an IRA may be preferable. The decision can be made on an individual basis, considering preoperative sphincter function, patient compliance, and risk of desmoid .
Desmoid Disease
Desmoids are locally aggressive but non-metastasizing myofibroblastic lesions which occur with disproportionately high frequency in patients with FAP . Putative risk factors include abdominal surgery, positive family history for desmoids, and site of the mutation (mutations beyond codon 1444). In contrast to sporadic desmoid tumors, the majority of the tumors associated with FAP are located in the abdominal wall or intra-abdominally. The tumors can be diagnosed by CT scanning or MRI. Desmoids occur most commonly in the peritoneal cavity (Fig. 52.8) and may infiltrate locally leading to small bowel, ureteric, or vascular obstruction. These lesions may progress rapidly or may resolve spontaneously, their unpredictable nature making them difficult to treat [32]. Attempted surgical resection carries a high morbidity and mortality and usually stimulates further growth. The options for treatment are pharmacological (nonsteroidal anti-inflammatory drugs (NSAIDs) [33] and/or anti-estrogens), chemotherapy, surgical excision, or radiotherapy. Pediatricians treating children with extraintestinal desmoid tumors should consider the possibility of FAP in the family .
Fig. 52.8
Intra-abdominal mass from a desmoid tumor seen in a teenager with FAP
Chemoprevention
NSAIDs may be protective against colon cancer by inhibiting prostaglandin synthesis via their effects on cyclooxygenase (COX) . Publications have shown a significant reduction in the number of rectal polyps in those patients taking the NSAID sulindac after colectomy. However, despite protracted drug use, the adenomas still progressed with case reports of rectal cancer. Sulindac administered before the development of polyps in genotype-positive adolescents did not prevent the development of adenomas.
Cyclo-oxygenase-2 (COX-2) enzyme is induced in inflammatory and neoplastic tissue. Selective COX-2 inhibitors generate fewer gastrointestinal-related side effects compared to the classical non-selective NSAIDs. One of these drugs (celecoxib) was found to reduce the number of colorectal adenomas by 28 % [34] and also to reduce the number of duodenal adenomas [35]. Unfortunately, cardiovascular side effects have been reported in patients using selective COX-2 inhibitors [36]. Although NSAIDs cannot replace surgical treatment for colonic FAP, they may play a role in postponing surgery in patients with mild colonic polyposis, or patients with rectal polyposis after prior colectomy . Other agents investigated but with so far inconclusive effect on polyp burden include: vitamin C, oral calcium, and ornithine decarboxylase inhibitor difluoromethylornithine (DFMO) and eicosapentaeoic acid present in fish oil [37, 38] .
Prognosis
Genetic investigations, colonic surveillance, and prophylactic colectomy have impacted favorably on mortality inFAP-affected patients . Studies that evaluated the mortality of patients with FAP reported that surveillance policies and prophylactic colectomy have resulted in a reduction in the number of FAP patients that died from CRC. Currently, a greater proportion of deaths are attributable to extracolonic manifestations of the disease (desmoid tumors, duodenal cancer). Central registration in a family cancer registry and prophylactic examination lead to a reduction of CRC-associated mortality and ensure appropriate follow up and patient support .
MYH-Associated Polyposis and Lynch Syndrome
MYH-associated polyposis (MAP) is characterized by the presence of adenomatous polyposis of the colorectum and an increased risk of CRC. Patientspresent with a variable number of polyps but no apparent extra-colonic features. This autosomal recessive condition results from a compound heterozygote of the mutY human homologue (MYH) gene located on chromosome 1p. MYH polyposis should have no pediatric implications as colonic polyposis typically occurs in patients in their 40s, although cancer and polyps can occur at earlier ages. Surveillance colonoscopy in affected individuals should commence at age 25 years.
Lynch syndrome is an inherited condition with a high penetrance for the development of early CRC caused by an alteration in DNA mismatch repair genes (MLH1, MSH2, MSH6, PMS2). With no described pediatric complications, the syndrome is associated with colorectal, endometrial, ovarian, gastric, renal tract, and other cancers. The median age for the development of CRC in Lynch syndrome is approximately 44 years and is uncommon below the age of 25 years. Revised criteria (Amsterdam II or Bethesda criteria) exist to help identify affected families and enable genetic testing for the DNA mismatch repair genes in early adulthood. Once identified as at risk, colonoscopic surveillance should be undertaken from the age of 20–25 years.
Other Polyposis Syndromes
Gorlin syndrome is an autosomal dominant condition comprising upper gastrointestinal hamartomas and pink or brown macules in exposed areas such as the face and hands.
Turcot’s syndrome (also referred to as brain tumor-polyposis syndrome) is characterized by concurrence of a primary brain tumor (most often glioblastoma multiforme) and multiple colorectal adenomas. The number of adenomas is often not high, but many of the reported patients have been adolescents . Patients with a polyposis syndrome and neurological symptoms should undergo thorough neurological examination and investigation for possible brain tumor.
In patients with long-standing inflammatory colitis, inflammatory polyps (pseudopolyps) may develop. Inflammatory polyps are of no significance and have no malignant potential.
The Role of a Polyposis Registry
There are numerous advantages to integrating a family into a cancer/polyposis registry . Polyposis registries are established across the world. A polyposis registry enables registration of polyposis patients and family members accessing counseling and genetic testing and initiation and coordination of screening of family members at risk of a polyposis syndrome. By increasing the rate of diagnosis of FAP and other polyposis syndromes, and enabling earlier diagnosis, there is proven improved survival of patients registered, almost certainly attributable to the improvement in organization and coordination of patient screening [39].
Other Tumors of the GI Tract (Excluding the Stomach and Hepato-biliary)
Gastrointestinal Stromal Tumors
Gastrointestinal stromal tumors (GISTs) are a unique variety of mesenchymal tumors that have recently been described. They are very uncommon among children and adolescents , and their true incidence is not well known due to the small number of described cases .
The overall incidence rates of GIST are reported to range between 6.5 and 14.5 per million per year [40–42]. The UK National Registry of Childhood Tumours showed an annual incidence of 0.02 per million children below the age of 14 years [43].
GISTs were described for the first time in 1990 and probably arise from neural interstitial cells of Cajal. Its molecular oncogenetics derives from mutations in the KIT oncogene (v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog) that encode for class III receptor tyrosine kinases and platelet-derived growth factor receptor alpha (PDGFRA), which are expressed in GIST patients. About 0–10 % of pediatric GISTs have an oncogenic KIT mutation [44–46], and only two patients were reported with a PDGFRA mutation to date [47, 48] which reflects probably other downstream activation pathway other than c-Kit mutation. A small subgroup of syndromic GISTs were found to have common germline mutations encoding the succinate dehydrogenase (SDH) gene [48, 49]. Recently, another molecule, IGF1R (insulin growth factor receptor 1), was found to be significantly expressed in a series of 17 GIST including one pediatric GIST, both by Western blot analysis and immunohistochemistry, and that it seems to be a new attractive pathway for this complex oncogenetic process [50].
Immunohistochemically, these tumors stain positive for vimentin (CD-117, a c-kit proto-oncogene protein) and CD-34—and stain negatively for smooth muscle actin [51]. Since the use of CD117 staining in 2002, 82 % of mesenquimatous tumors were considered to be GISTs, so its’ true incidence rose since there, with a proportional decline of smooth muscle tumors cases [52]. Histologically, GISTs are composed either of spindle-shaped cells, epithelioid cells, or a combination of both, being spindle-shaped type slightly more frequent in a large analysis of 99 of 113 pediatric cases [53].
GISTs can present as sporadic or inherited cases, and the latter can be inherited through familial autosomal dominant fashion or as syndromic tumors, named as the Carney triad.
Carney Triad
GIST can occur rarely in association with other tumor syndromes. The association of gastric leiomyosarcoma, extra-adrenal paraganglioma and pulmonary chondroma was first described in 1977 and subsequently named the Carney triad [54]. It affects predominantly females, and it has been described as a chronic benign-course tumor that comprises recently also adrenocortical tumors and esophageal leiomyoma [55–57]. Neither KIT, PDGFRA, nor SDH mutations were found in patients with the Carney triad. In 2002, Carney and Stratakis distinguished an inherited tumor syndrome comprising GIST and paragangliomas which have common germline mutations encoding SDH subunits B, C, D, and it was termed Carney–Stratakis diad [58] .
Familial
Familial GIST cases are very rare, and they are autosomal dominant inherited. They differ from Carney dyad as they are heritable germline KIT mutations tumors. Occurrence of multiple tumors can be observed both in syndromic [59–64] and in sporadic GIST. Familial GISTs do not have female predominance, and the main location is small intestine, in contrast to sporadic GIST where the stomach is the main affected place [61, 65]. Other phenotypic characteristics associated with familial GIST include mastocytosis [66], dysphagia [67, 68], cutaneous hyperpigmentation or urticaria pigmentosa [59, 61, 64, 69, 70], and recurrent small intestinal diverticular perforation [68]. Familial GIST does not develop early in life, as the youngest patient in a GIST family described to this date had symptoms at 18 years old [70] .
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


