Polyposis and Hereditary Cancer Syndromes
Polyposis Syndromes
Introduction
Polyps develop throughout the gastrointestinal (GI) system either as sporadic lesions or as part of a polyposis or hereditary cancer syndrome. The most common syndromes are those that involve neoplastic intestinal adenomas. These include familial adenomatous polyposis, MYH polyposis, and hereditary nonpolyposis colorectal cancer syndrome. Less common polyposis syndromes involve development of hamartomas and include Peutz-Jeghers, juvenile polyposis, Bannayan-Riley-Ruvalcaba, neurofibromatosis 1, and Cowden syndromes. Another hamartomatous polyposis syndrome, Cronkhite-Canada syndrome, is nonfamilial. Other syndromes, such as hyperplastic polyposis and hereditary mixed polyposis, include both adenomatous and nonadenomatous polyps. Some rare forms of polyposis involve proliferations of lymphoid or mesenchymal tissues and are discussed elsewhere in this text.
Less than 1% of all GI malignancies result from intestinal polyposis syndromes. One classification scheme of polyposis syndromes is shown in Table 12.1. In order to diagnose a specific polyposis syndrome, one must be aware of (a) the number of polyps a patient has and their location, (b) the patient’s age, (c) the family history, and (d) other clinical information that identifies a patient as having a specific syndrome. Overlap exists between several syndromes.
Hereditary Adenomatous Polyposis Syndromes
Familial Adenomatous Polyposis and Its Variants
General Comments
Familial adenomatous polyposis (FAP) (OMIM entry #175100) is a generalized growth disorder that includes intestinal polyposis as well as numerous extracolonic manifestations. Overlap exists among the different adenomatous polyposis syndromes, and the varied extraintestinal manifestations of FAP have led to confusion in their nomenclature. Gardner syndrome was the term applied to patients with colonic polyposis, epidermoid cysts, osteomas, and desmoids. Turcot syndrome was diagnosed if the patient had colonic polyposis with brain tumors. However, our current understanding of these disorders indicates that FAP, Gardner syndrome, and Turcot syndrome all represent variations of the same disease rather than distinct entities, and result from defects in a single pleiotropic gene with variable expressivity.
Genetic Features
The FAP-associated gene is located on chromosome 5, and is called APC for adenomatous polyposis coli (1,2,3,4). APC has 15 exons (Fig. 12.1) and encodes a protein of 2,843 amino acids (2,4). The large size of the gene may account for the high frequency of new mutations that occur in it. Germline mutations in APC account for most cases of FAP (2,3,5). Every cell in an FAP patient contains one inactive APC allele; an alteration in the other allele gives rise to intestinal tumors. Inactivation of the second allele occurs in the earliest recognizable phase of the tumors, including some lesions containing as few as two adenomatous crypts, confirming that inactivation of the second APC allele occurs early in neoplastic development.
APC germline mutations are notable in that they are almost exclusively single base pair (bp) changes leading to termination codons, or small (one to four bp) deletions, insertions, or splicing mutations causing translational frame shifts and subsequent downstream stop codons (2,3,6). Even though 45% of patients have detectable germline defects in exon 15, mutations are located throughout the length of the APC gene. Most mutations disrupt the coding sequence in the 5’ half of the FAP open reading frame (7).
The first clue to the function of the APC protein came when immunoprecipitation experiments showed that anti-APC antibodies precipitated β-catenin (8). APC and β-catenin are part of a complex signaling pathway, the Wnt pathway, that controls many cellular processes including proliferation, differentiation, apoptosis, and body patterning during development (Fig 12.2). In addition to its role in mediating β-catenin degradation, APC also plays a part in controlling the cell cycle, inhibiting progression from the G0/G1 phase of the cycle to the S phase (9). APC also
stabilizes microtubules, promoting chromosomal stability. Loss of APC function results in defective mitotic spindle formation and abnormal chromosomal segregation (10).
stabilizes microtubules, promoting chromosomal stability. Loss of APC function results in defective mitotic spindle formation and abnormal chromosomal segregation (10).
TABLE 12.1 Classification of Intestinal Polyposes | |||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||
Disease Expression
Patients with FAP exhibit considerable phenotypic variability of their colonic and extracolonic lesions, even within the same family. This suggests that the nature of the inherited genetic defect is only one parameter determining patient phenotype. Inactivation of APC may provide affected cells with a proliferative growth advantage (6), allowing the colon to then acquire additional genetic abnormalities, thereby facilitating disease progression. Environmental factors may also be important. Bile from FAP patients appears to be more mutagenic than that from non-FAP patients (11), perhaps inducing secondary changes both in APC and other genes in the adenoma–carcinoma sequence (see Chapter 14). Many manifestations of FAP may be partially controlled by hormonal status and other genetic factors. Data supporting such a postulate include (a) stimulation of polyp growth during puberty, (b) adenoma inhibition by sulindac (12), (c) the preponderant development of thyroid carcinomas in women (13), and (d) the association of repeated trauma with the development of desmoid tumors (see Chapter 19).
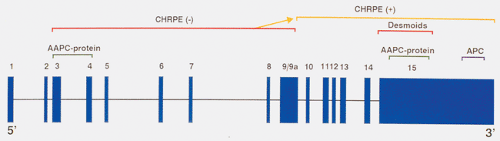 FIG. 12.1. The APC gene with its 15 exons shown as blue boxes. Mutations occurring within the part of the gene designated by the red line result in a congenital hypertrophy of the retinal pigment epithelium (CHRPE)-negative phenotype. Those occurring in the area designated by the yellow line are associated with CHRPE lesions. The yellow arrow indicates the region of the gene in which mutations begin to result in the appearance of CHRPE lesions. The orange area of the gene is associated with formation of desmoid tumors. Mutations in the green and purple areas result in the attenuated APC and full-blown APC phenotypes, respectively. AAPC, attenuated adenomatous polyposis coli. |
Relationship of APC Mutations to Disease Expression
Mutations in specific regions of APC produce different clinical phenotypes, and the length of the truncated gene product influences the severity of the colonic disease and the presence of eye lesions or desmoid tumors (Fig. 12.1). Patients with mutations toward the 3’ end of exon 15 who produce longer protein products have a more severe phenotype than patients with mutations toward the 5’ end of the gene (exons 3 and 4). Patients with less severe forms of the disease are referred to as having attenuated adenomatous polyposis (discussed below). Patients with the severe form of the disease tend to have large numbers of polyps that arise early in life (14,15,16). The commonly observed deletion of five
bp at codon 1309 within exon 15 associates with early onset of colon cancer (1) and the early development of colonic adenomas.
bp at codon 1309 within exon 15 associates with early onset of colon cancer (1) and the early development of colonic adenomas.
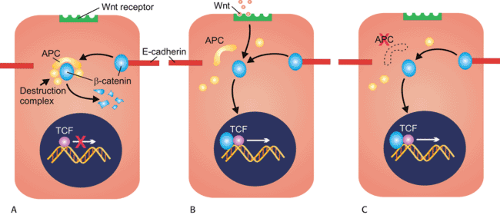 FIG. 12.2. Diagram of the Wnt signaling pathway. A: In normal cells, most endogenous β-catenin is bound to the intercellular adhesion molecule E-cadherin on the cell membrane. Any unbound, cytoplasmic β-catenin is quickly targeted for degradation by a protein complex that includes the proteins Axin, protein phosphatase 2A, protein kinases GSK3β and CK1α, and APC. Phosphorylation of β-catenin by this complex targets it for ubiquitin-mediated destruction. B: Stimulation of the wnt receptor by its ligand results in the destruction protein complex, which is inactivated, allowing cytoplasmic β-catenin to enter the nucleus and interact with the Tcf family of transcription factors. The result is expression of WNT target proteins and increased cell proliferation. C: In patients with familial adenomatous polyposis, the dysfunctional APC protein disrupts the activity of the multisubunit destruction complex, and results in accumulation of free β-catenin in the cell. β-Catenin is free to enter the nucleus of the cell where it interacts with Tcf, resulting in sustained expression of c-Myc, cyclin D1, and other Wnt target proteins. |
Attenuated Adenomatous Polyposis
A less severe form of FAP, known as attenuated adenomatous polyposis coli (AAPC), is characterized by a relatively low number of adenomas (15). AAPC is associated with a germline APC mutation in approximately 10% of affected patients (17). This syndrome differs significantly from classic FAP in that affected individuals have fewer adenomas that tend to cluster on the right side of the colon where they appear flat rather than polypoid. The number of adenomas varies among family members, ranging from 1 or 2 to 100. Upper gastrointestinal lesions, particularly fundic gland polyps, are almost invariably present. In addition, affected patients exhibit a reduced risk for colorectal cancer compared with those with classic FAP.
The lifetime penetrance of colon cancer is high in AAPC families, however, even among members with relatively few adenomas. When colorectal cancers do develop, they arise later than in classic FAP. The average age of colon cancer onset in AAPC patients is approximately 15 years later than that of patients with classic FAP and approximately 10 years earlier than individuals with sporadic colorectal cancer. Table 12.2 compares classic FAP and AAPC.
Clinical Features
FAP is the most frequent genetic polyposis syndrome, affecting 1 in 6,850 to 29,000 live births. It is transmitted as an autosomal dominant disorder with up to 90% penetrance. Fifty percent of the children of an affected individual and a normal individual will inherit the polyposis gene and will develop the disease. Ten to thirty percent of patients with FAP have no familial history and represent spontaneous new mutations. FAP patients develop at least a few adenomas by age 21. The adenomas almost always involve the rectosigmoid, where they tend to develop rapidly and in large numbers. Males and females are equally affected (18).
As noted above, variation in the expression of the FAP phenotype within families exists, and may result from dietary or environmental factors. Even more variation exists between families, presumably due to the fact that APC mutations occur at multiple sites in the gene and that different mutations confer different phenotypes.
Adenomas are not present at birth in FAP patients. Most affected individuals remain asymptomatic until puberty, at which time the polyps begin to appear (19). In untreated,
unscreened patients, the mean age of polyp development is 24.5 years, symptom onset is 33 years, polyp diagnosis is 35.8 years, cancer diagnosis is at 39.2 years, and death from cancer averages a mean of 42 years (19). Young patients present with a small number of polyps, the number of which progressively increases with time. Eventually, the entire length of the colon becomes carpeted with adenomas. By the time a patient comes to colectomy, he or she may have hundreds to tens of thousands of polyps. Progression to cancer is inevitable; by age 30, approximately 75% of FAP patients will have developed colon carcinoma unless a prophylactic colectomy is performed. Most untreated patients die of cancer by the 5th decade of life.
unscreened patients, the mean age of polyp development is 24.5 years, symptom onset is 33 years, polyp diagnosis is 35.8 years, cancer diagnosis is at 39.2 years, and death from cancer averages a mean of 42 years (19). Young patients present with a small number of polyps, the number of which progressively increases with time. Eventually, the entire length of the colon becomes carpeted with adenomas. By the time a patient comes to colectomy, he or she may have hundreds to tens of thousands of polyps. Progression to cancer is inevitable; by age 30, approximately 75% of FAP patients will have developed colon carcinoma unless a prophylactic colectomy is performed. Most untreated patients die of cancer by the 5th decade of life.
TABLE 12.2 Comparison of FAP and AAPC Colorectal Cancer Syndromes | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
Adenomas also develop in extracolonic sites, most commonly in the duodenum around the ampulla of Vater. Patients less frequently develop adenomas in the stomach or in other portions of the small intestine. Carcinomas may subsequently arise in these sites, but are relatively rare. Gastric cancers are more common in FAP patients living in parts of the world where gastric cancer rates are high. Fifty percent of Japanese FAP patients develop gastric adenomas, and gastric carcinoma is more common in these patients than ampullary cancers. In contrast, Western gene carriers exhibit a higher rate of ampullary than gastric neoplasms (20).
Colonic adenomas are often present for years before giving rise to symptoms including rectal bleeding, colicky abdominal pain, diarrhea, and mucous discharge (19). Seventy-five percent of polyposis patients without cancer have rectal bleeding, and 63% have diarrhea (19). When symptoms are severe enough to cause concern, two thirds of the patients have already developed a carcinoma. Very rarely, patients develop severe electrolyte depletion as a result of diffuse polyposis (21). Acute pancreatitis develops secondary to obstruction of the pancreatic duct at the ampulla of Vater by adenomas. Intussusception due to the adenomas may also occur.
Diagnosis of Familial Adenomatous Polyposis
The diagnosis of symptomatic patients is usually not difficult because they present with bleeding, diarrhea, or abdominal pain. Surveillance of children in affected kindreds results in earlier adenoma detection and in a lower cancer incidence. Today the tendency is to diagnose the presymptomatic individual by genetic testing and then confirm the diagnosis by sigmoidoscopy. Some advocate beginning flexible sigmoidoscopy by age 10 to 12 and continued yearly examinations until age 35 (22). Upper GI endoscopy should begin once the
diagnosis of FAP is made and should be continued every 3 years thereafter in the absence of gastroduodenal adenomas (23). Genetic techniques will detect 50% of carriers of APC mutations (24).
diagnosis of FAP is made and should be continued every 3 years thereafter in the absence of gastroduodenal adenomas (23). Genetic techniques will detect 50% of carriers of APC mutations (24).
The child of an FAP patient has a 50% likelihood of inheriting the genetic mutation. Prescreening strategies can be designed to detect mutations at the 12 most commonly mutated loci, which account for nearly 40% of germline mutations in FAP patients (25). The use of DNA extracted from archival specimens of affected available FAP patients increases the number of at-risk individuals who can be diagnosed presymptomatically (25). The association of the DNA markers and the presence of congenital hypertrophy of the retinal pigment epithelium (CHRPE) make it possible to identify gene carriers even in the absence of rectal polyps. CHRPEs affect up to 90% of classic FAP patients, especially those with both upper gastrointestinal and extraintestinal manifestations (26). They may be single or multiple, bilateral or unilateral, and are easily identified on funduscopic examination. These congenital lesions are observed in very young or even preterm infants, and represent the earliest diagnostic stigma of FAP. If one family member has CHRPE, all family members with the disease will have CHRPE. Similarly, other kindreds exist in which no family members have the retinal lesions.
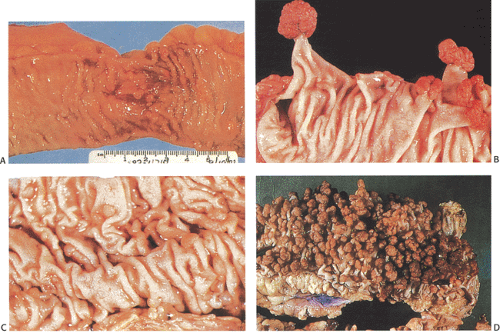 FIG. 12.3. Gross features of familial polyposis. A through D demonstrate the different forms this disorder may take. A: Numerous sessile, small rounded polypoid lesions are present, often on the mucosal folds. Large intervening areas of apparently normal colon are present. B: Clusters of pedunculated adenomatous polyps, larger than those seen in A, are present. They tend to pull up the mucosal folds into stalks. C: Numerous sessile and pedunculated polyps are scattered over the mucosal surface. D: The bowel is carpeted by numerous larger raspberrylike pedunculated polyps. |
Cancer Development
Carcinoma invariably develops in FAP patients if the colon is not resected by age 40 or 50. Indeed, FAP is the “experiment of nature” that provides support for the colonic adenoma–carcinoma sequence (Fig. 12.3). Adenomas also represent the precursor for small intestinal cancer, but small intestinal adenomas are less likely to become malignant than are colonic lesions. Adenomas and carcinomas also develop in the retained rectum following colectomy.
Carcinomas develop approximately 6 years after symptom onset. The incidence of carcinoma is approximately 10% in
patients observed for 5 years and 50% in those observed for 20 years. Each 10-year age group has a 2.4-fold increase in cancer risk (27). Multiple cancers are frequent, with synchronous lesions affecting 41% of patients and metachronous lesions affecting 70%. Polyp count and patient age, but not sex, predict cancer risk. Patients with >1,000 polyps have a 2.3 times greater risk of cancer than those with <1,000 polyps.
patients observed for 5 years and 50% in those observed for 20 years. Each 10-year age group has a 2.4-fold increase in cancer risk (27). Multiple cancers are frequent, with synchronous lesions affecting 41% of patients and metachronous lesions affecting 70%. Polyp count and patient age, but not sex, predict cancer risk. Patients with >1,000 polyps have a 2.3 times greater risk of cancer than those with <1,000 polyps.
Patients who undergo prophylactic colectomy may still die of other tumors, including ampullary cancers, brain tumors, hepatoblastomas, and desmoid tumors (28).
Treatment
Because of the high cancer risk, FAP patients undergo prophylactic total colectomy once the diagnosis is established. The procedure is timed so as not to interfere with psychosocial development, if the individual is a teenager or younger. It is recommended that the procedure be performed by age 20 to 25 (29). Patients have four surgical options: Total proctocolectomy, continent ileostomy (Koch pouch), total abdominal colectomy with ileorectal anastomosis, and ileoanal reservoir procedure. Total proctocolectomy with ileostomy eliminates the possibility of rectal carcinoma, but this procedure is unacceptable to many patients. The ileoanal reservoir procedure removes the bulk of the colorectal mucosa at risk for developing cancer while preserving the anal sphincter, but the cancer risk continues in the retained rectum. Rectal preservation is an ideal therapy for patients with relatively few rectal polyps because such patients are less likely to develop rectal carcinoma than those with numerous rectal adenomas (30). Patients with a retained rectum undergo semiannual screening with removal of all polyps that develop. According to some, more than 50% of patients will develop carcinoma in the rectal stump despite semiannual sigmoidoscopic surveillance and removal of all polyps (30).
Adenoma and Carcinoma Distribution
Adenomas develop throughout the entire colon (Fig. 12.4) and appendix. They are fairly evenly distributed throughout the large intestine, with a tendency for them to be larger in the sigmoid and the rectum, thereby making the density appear greater in this region (19). Rarely, the rectum is spared, especially in AAPC. When extensive, the entire large bowel becomes carpeted with adenomas (Fig. 12.3). Adenomas show gradations in size and shape from pedunculated tumors 1 cm or more in diameter, to smaller, broader-based nodules, to tiny lesions 1 mm or less in size. Adenomas tend to be larger in propositi (Figs. 12.3, 12.4, and 12.5) than in patients undergoing screening surveillance (Fig. 12.5). In classic FAP, the number of polyps ranges from <100 (Fig. 12.3) to >5,000 with an average of 1,000, depending on when one sees the patient (19). Colorectal carcinomas may be multifocal and more frequently develop on the left side of the colon (33). Patients with AAPC develop flatter, nonpolypoid adenomas than are seen in classic FAP. They arise throughout the colon, with preferential involvement of the right colon. They also originate in the retained rectum following colectomy.
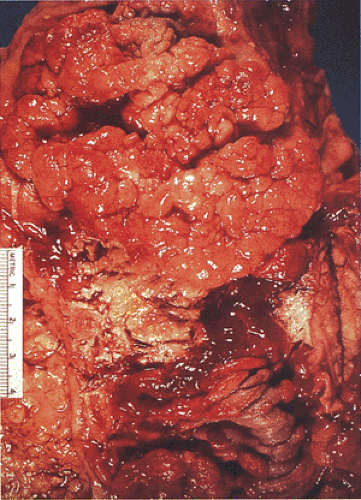 FIG. 12.4. Resection specimen in a familial adenomatous polyposis patient. There is a large fungating tumor present that represented an invasive carcinoma. Numerous pedunculated adenomas are seen in the surrounding mucosa. |
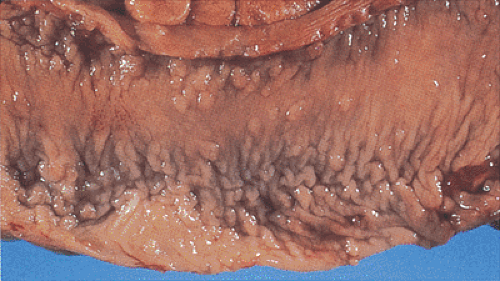 FIG. 12.5. Resection specimen from a patient who was part of a surveillance program. The patient has numerous small nodular lesions only mildly elevated over the mucosal surface. The lesions are smaller in patients undergoing surveillance than those who are not part of surveillance programs. |
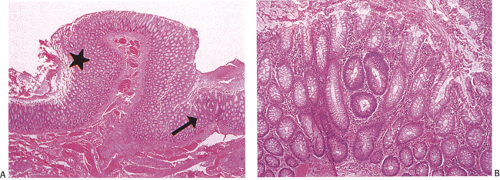 FIG. 12.6. Cross sections of the grossly apparently normal mucosa in patients with familial polyposis. A: Low-power picture showing two foci containing adenomatous glands. A unicryptal adenoma is present above the star. A tricryptal adenoma is illustrated by the arrow. B: A tricryptal adenoma is illustrated. |
Pathologic Features of Adenomas
Adenomas and carcinomas in FAP patients grossly resemble their sporadic counterparts. Endoscopically, very small adenomas resemble hyperplastic polyps (Figs. 12.3 and 12.5). It is only when they become larger that the typical raspberrylike configuration of an adenoma becomes evident. As in sporadic colon cancer, the incidence of malignancy relates to adenoma size.
In the early stages, adenomas consist of small groups of adenomatous tubules. They range from unicryptal, bicryptal, or tricryptal lesions in grossly normal-appearing mucosa (Figs. 12.6 and 12.7) to the more typical multicryptal grossly visible polyps seen in patients without FAP (Fig. 12.8). The presence of unicryptal, bicryptal, and tricryptal adenomas strongly suggests the diagnosis of FAP. Even in unicryptal adenomas, the entire tubule is completely lined by neoplastic epithelium (Fig. 12.7). Proliferation throughout the entire length of the adenomatous crypt leads to branching, budding, infolding, and mucosal elevation. As the lesions enlarge to a grossly visible size, they become tubulovillous. Pure villous adenomas are rare in FAP patients.
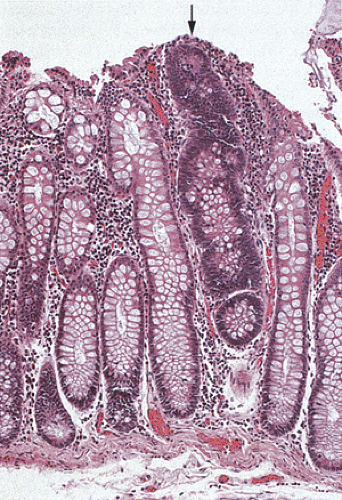 FIG. 12.7. Longitudinal section of the mucosa showing the presence of a unicryptal adenoma (arrow). Even the single adenomatous crypts are easily visible. |
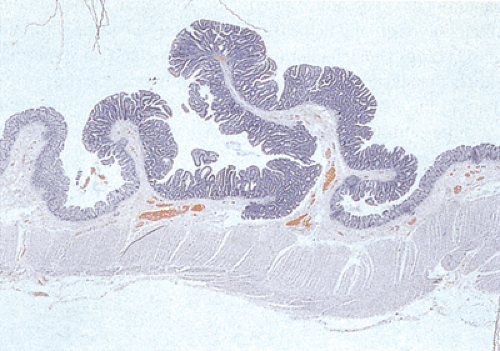 FIG. 12.8. Familial polyposis. Histologic section through the mucosa demonstrating the presence of multiple adenomatous polyps arising on the surface of the mucosa. |
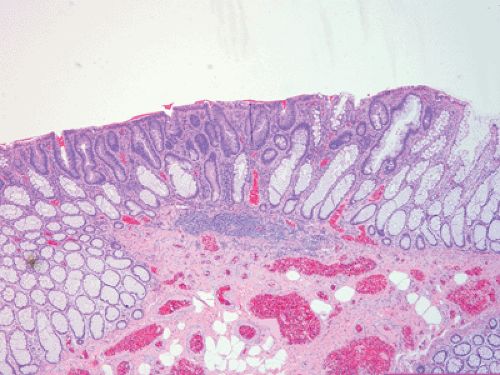 FIG. 12.9. Flat adenoma in a patient with attenuated adenomatous polyposis coli. Note that the adenomatous glands (center) lie at or below the level of the normal mucosa. |
FAP patients develop depressed, flat, or polypoid adenomas (34), with AAPC patients showing a tendency to develop flat lesions. In contrast to pedunculated adenomas, the whole surface of flat or depressed adenomas lies at or below the level of the normal mucosa (Fig. 12.9). Polypoid adenomas are those with convex surfaces. Flat adenomas differ endoscopically and histologically from the usual adenoma. They present as slightly elevated plaques of adenomatous mucosa, not more than twice as thick as the adjacent normal mucosa. Further growth is by radial extension of adenomatous epithelium so that the lesions remain flat. When cancer develops, then one sees a reddish depression surrounded by marginal elevations (35). Occasionally, patients with FAP develop serrated polyps (see Chapter 14) or, more rarely, they develop juvenile polyps.
Upper Gastrointestinal Lesions
Nearly all FAP patients have polyps in the upper GI tract, with as many as 90% of patients developing gastric or duodenal adenomas by age 70 (36). Adenomas develop in the gastric antrum, duodenum, periampullary region, and ileum. However, it is the periampullary region that is most commonly involved, and adenomas tend to cluster at this site. More than 50% of FAP patients who undergo upper endoscopy have a grossly polypoid lesion, 90% of which arise in the periampullary region (37), suggesting that bile plays a role in their growth (38). The bile of FAP patients has a greater proportion of chenodeoxycholic and a lower proportion of deoxycholic acid than does the bile of patients without polyposis (38) and is more mutagenic. Patients also exhibit fecal flora abnormalities resulting in the possible production of carcinogenic compounds.
Periampullary carcinoma is a major cause of death in FAP patients (39), affecting from 2.9% to 12% of all FAP patients (19,39,40) and causing death in 22.2% of patients following colectomy (40).
Duodenal Lesions
Duodenal adenomas develop in as many as 100% of patients in Japanese series (40,41) and in 50% in Western countries (19,42,43). Duodenal adenomas develop when patients are in their 2nd to 5th decades of life. The average age of FAP patients with adenomas involving the ampulla of Vater is 31.7 years, compared with 59.6 years in those without FAP. Duodenal adenomas vary in size and appearance from microadenomas in a normal-appearing ampulla to sessile polyps measuring 3 cm in diameter (43). Duodenal adenomas are generally small in screened populations. Over 90% of duodenal adenomas are tubular lesions. Large lesions become tubulovillous with dysplasia ranging from mild to moderate in degree. FAP-associated duodenal adenomas show a significant increase in the number of Paneth cells (Fig. 12.10) and endocrine cells per crypt compared to controls. This specialized cell hyperplasia affects the flat mucosa of FAP patients, regardless of the presence or absence of adenomas, and may represent a primary defect in the regulation of duodenal stem cell differentiation in FAP patients (44).
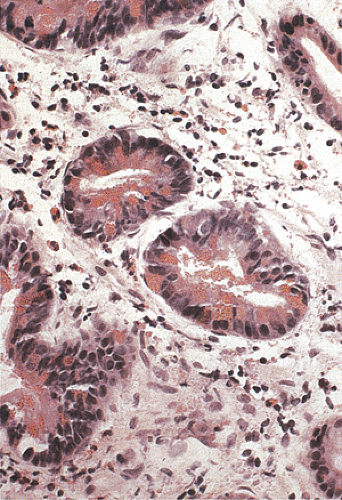 FIG. 12.10. Duodenal adenoma in familial adenomatous polyposis patient showing the presence of adenomatous epithelium and a large number of Paneth cells, as evidenced by the eosinophilic granules within the cytoplasm. |
Ileal and Jejunal Lesions
Adenomas also develop in the ileum and jejunum, but to a lesser extent than in the duodenum. As many as 82% of FAP patients develop ileal adenomas (45). Ileorectal anastomoses, ileostomies, and ileal pouches predispose the ileal mucosa to become neoplastic (32,46,47). The ileal mucosa undergoes colonic metaplasia, which then gives rise to adenomas. Ileal adenomas resemble duodenal, gastric, and large intestinal adenomas. Ileal adenomas tend to be sessile, measuring 1 to 5 mm in size. Multiple lymphoid polyps also develop in the terminal ileum in FAP patients.
Gastric Lesions
Gastric polyps develop in approximately two thirds of FAP patients (23). Two different types of polyps arise in the stomach. Antral polyps are usually adenomas, whereas the small polyps arising in the fundus and body are usually fundic gland polyps (see Chapter 4) (48). Fundic gland polyps affect 25% to 60% of FAP patients.
Fundic gland polyps occur earlier than gastric adenomas, presumably because they originate in the existing fundic mucosa without the requirement for intervening intestinal metaplasia. Most patients with fundic polyps are under age 20. Fundic gland polyps are often multiple and small in diameter, appearing sessile or semi-sessile. They are identical to sporadic fundic gland polyps (see Chapter 4) (Fig. 12.11). The gastric mucosa may be studded with numerous small, sometimes eroded polyps that may increase in number and size over a several-year period. Alternately, they may decrease, or even disappear. Lesions that decrease or disappear may be followed by the appearance of a new crop of polyps (48). The fundic gland lesions are generally considered to be benign with little or no malignant potential, yet dysplasias and carcinomas have been described in FAP-associated fundic gland polyps (49,50). Superimposed gastric adenomas may give the false impression of dysplasia arising in a fundic gland polyp.
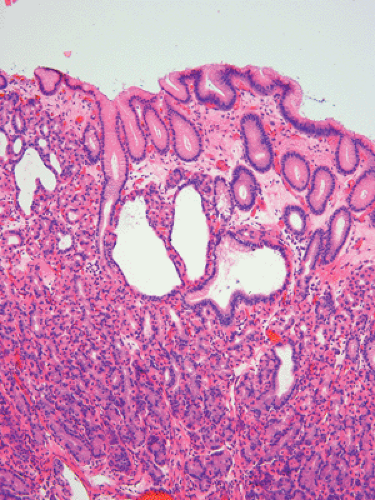 FIG 12.11. Fundic gland polyp in a patient with attenuated familial adenomatous polyposis. The lesion is similar in appearance to a sporadic fundic gland polyp. |
In Western countries, FAP-associated gastric adenomas and carcinomas are rare, contrasting with the Japanese experience, and supporting the role of environmental or other genetic factors in gastric cancer development. Gastric adenomas develop in the antrum in areas of intestinal metaplasia, a histologic requirement for the formation of gastric adenomas. When one compares gastric adenomas with colonic adenomas, the gastric lesions tend to be smaller and more sparsely distributed. In addition, gastric adenomas occur later in life than colonic adenomas. Gastric adenocarcinomas develop in the adenomas. Finally, FAP patients may develop gastric microcarcinoid tumors (48).
Adenomas and Carcinomas in the Rectal Remnant
Overall, the cumulative risk of developing cancer in the retained rectum ranges from 4% to 59% during a period of 10 to 30 years following surgery (51,52). The cancers may be small, depressed, and restricted to the mucosa. The age of the patient at the time of colectomy, the length of the retained colon, the tendency for spontaneous regression of polyps in the retained rectum, and the presence of carcinoma in the excised colon all influence the subsequent development of rectal cancer. Carcinomas and adenomas can develop, even in patients who are closely followed (32).
Kinetic and Other Biologic Abnormalities
FAP patients have a generalized abnormality of DNA synthesis that eventually results in uncontrolled cellular replication in the GI tract and in extragastrointestinal sites. FAP patients often exhibit increased ornithine decarboxylase activity, an enzyme that is essential for intestinal mucosal proliferation (53). As a result, FAP patients demonstrate increased cell proliferation in the adenomatous epithelium as well in the nonneoplastic crypts, on the luminal surface, and in the mucosa between adenomatous polyps (54). The markedly increased replication rate and the large number of adenomatous cells available for mutation and malignant transformation increase the likelihood of cancer developing.
APC mutation is a requisite initiating event for the formation of adenomas and provides a permissive environment for the subsequent development of carcinomas. Further steps in the neoplastic progression, including severe dysplasia and invasive carcinoma, associate with somatic mutations
in K-ras and p53 (55,56). In addition to mutations, losses of the APC, p53, and DCC also occur (56,57,58). Additionally, c-myc is frequently overexpressed (59). Some patients also develop microsatellite instability (60).
in K-ras and p53 (55,56). In addition to mutations, losses of the APC, p53, and DCC also occur (56,57,58). Additionally, c-myc is frequently overexpressed (59). Some patients also develop microsatellite instability (60).
Extraintestinal Manifestations
FAP patients have a high incidence of extraintestinal manifestations, including dental and skin abnormalities and the development of various types of neoplasms (Fig. 12.12). The dental abnormalities include unerupted teeth, supernumerary teeth, dentigerous cysts, and mandibular cysts. Subcutaneous lesions include epidermoid cysts, lipomas, fibromas, neurofibromas, and trichoepitheliomas. The latter appear at an early age, even before polyps appear. Those that occur in prepubertal years are strong indicators for the presence of a polyposis syndrome, and to some represent an indicator for regular sigmoidoscopy, even without a history of polyposis. Epidermal inclusion cysts are often multiple. The epidermoid cysts occur anywhere on the body but most are located on the arms, buttocks, legs, and face, and occasionally in the testis.
Not surprisingly, patients who carry germline mutations in a tumor suppressor gene exhibit tumors in sites other than the GI tract. These are summarized in Figure 12.12. Osteomas commonly occur in the skull or jaw, although they can affect any bone. These benign tumors rarely become malignant. FAP associates with nasopharyngeal angiofibromas (61). The lesion occurs 25 times more commonly in FAP patients when compared with the general population.
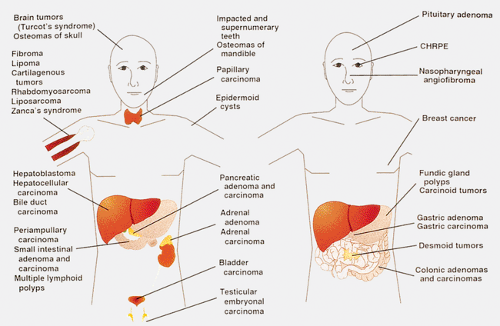 FIG. 12.12. Diagram of the various manifestations of familial adenomatous polyposis, which is essentially a systemic disorder. CHRPE, congenital hypertrophy of the retinal pigment epithelium. |
FAP patients also develop a number of endocrine and other neoplasms. Thyroid carcinomas, which occur with increased frequency in FAP patients, are all follicular neoplasms, sometimes containing papillary, cribriform, solid, and spindle cell components. FAP-associated thyroid cancers are commonly multifocal and predominantly affect young women. Since multicentricity is unusual for follicular thyroid tumors, it should alert the pathologist to the possibility of FAP (62). Soft tissue lesions include fibromas, lipomas, and desmoid tumors.
Desmoid tumors are a locally invasive form of fibromatosis (see Chapter 19) that affects 9% to 32% of FAP patients (63,64), particularly women (3:1). Affected patients are often young, with a mean age of 29.8 years (65). The overall prevalence of these lesions in FAP is 15%, a risk approximately 850 times greater than that of the general population (65,66).
Desmoid tumors tend to involve members of the same family and associate with mutations in exon 15 of APC (Fig. 12.1). Most patients have undergone a previous colectomy
(86%). Hormonal factors such as pregnancy and estrogen use may also play etiologic roles in desmoid development. Although the desmoids can develop on the shoulder girdle, buttocks, groin, and abdominal wall, the majority arise within the small intestinal mesentery.
(86%). Hormonal factors such as pregnancy and estrogen use may also play etiologic roles in desmoid development. Although the desmoids can develop on the shoulder girdle, buttocks, groin, and abdominal wall, the majority arise within the small intestinal mesentery.
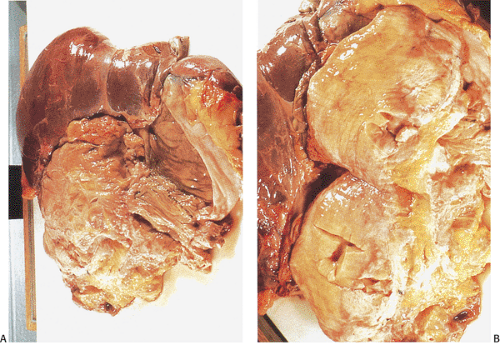 FIG. 12.13. Desmoid tumor in a patient who died of complications from the desmoid. A: Autopsy en bloc resection of the liver, spleen, intestines, and desmoid tumor. One can see that the tumor diffusely infiltrates the abdominal contents and extends up to the liver. Numerous organs were entrapped within this neoplasm, including the biliary tree, large numbers of vessels, and loops of bowel. B: Higher magnification of the neoplasm showing the presence of a fleshy mass with areas of ischemic necrosis. |
Desmoid tumors represent an adverse prognostic factor in FAP patients because they associate with a high frequency of complications and tumor recurrence. These nonencapsulated, irregular, infiltrative (Fig. 12.13), locally aggressive lesions do not metastasize, but they can cause significant intestinal obstruction, ureteral or vascular compression, or other local problems. Extensive mesenteric or retroperitoneal involvement leads to recurrent small bowel obstruction. Many patients die from these lesions. In one center, desmoid tumors were second only to colorectal cancer as a cause of death in FAP patients (39). Death results from vascular compromise, small bowel gangrene, perforation, or intra-abdominal sepsis.
Surgery is usually reserved for a specific indication, such as relief of intestinal or ureteric obstruction. Surgery on these lesions is extremely difficult because it is rare for the tumors to be small enough or sufficiently localized to allow resection without sacrificing vital structures. In most cases, attempts at surgical resection are followed by tumor recurrence. The recurrence rate after wide surgical resection is 81% (63). As a result, some have abandoned surgical procedures in favor of other therapies.
Other therapies have been tried including radiotherapy and various chemotherapies. Radiotherapy slows or reverses abdominal wall and mesenteric tumor growth, but the dose is limited by the need to preserve underlying intraperitoneal structures. Hormonal therapy, especially with antiestrogens, has met with minimal success. Tamoxifen has been effective in some patients (67), as has progesterone (68). Recently, NSAIDs have been used to halt the tumor growth, presumably by interfering with prostaglandin metabolism. Treatment with sulindac has resulted in a reduction in tumor size in some patients, but the response is inconsistent (67).
Histologically, desmoids consist of uniform, mature fibroblasts arranged in intertwining bundles. Mitoses are infrequent and never atypical. The extent of vascularization varies and may be prominent (Fig. 12.14). This prominent vascular ectasia, which sometimes occurs in FAP patients, is not a feature in non-FAP individuals and may account for intraoperative hemorrhagic complications. The tumor infiltrates the intestinal loops and peritoneum. The arteries and veins become surrounded by tumor but it does not infiltrate them (Fig. 12.14). The tumor cells actively produce collagen fibers.
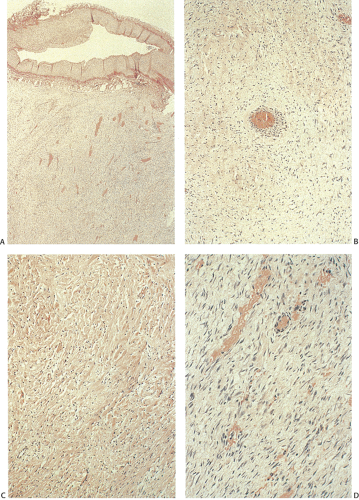 FIG. 12.14. Desmoid tumor. A: A highly vascular spindled cell mass is present, which extends up to a large sclerotic vessel but does not invade it. B: Higher magnification showing the haphazard arrangement of the spindled cells. C: A more hyalinized area of the tumor. D: A more vascular portion of the tumor. |
TABLE 12.3 Central Nervous System Lesions in Turcot Syndrome | |
|---|---|
|
Turcot Syndrome
Turcot syndrome is the association of FAP with malignant tumors of the central nervous system. Central nervous system tumors arise around the time of puberty, often before the diagnosis of FAP (Table 12.3). The brain tumors are often lethal (71). Both familial and nonfamilial cases occur, resulting in a controversy concerning its mode of inheritance. Some suggest an autosomal recessive inheritance pattern (72,73). However, patients often die before having children, making it difficult to test the inheritance pattern. Lewis et al proposed classifying Turcot syndrome based on a study of family pedigrees (74) (Table 12.4).
Patients with Turcot syndrome fall into three groups based on their intestinal lesions: (a) those with a low number of colonic polyps (20 to 200), (b) those with large polyps measuring over 3 cm in diameter, and (c) those with colonic carcinoma developing during the 2nd to 3rd decades. Patients in the second group often have very few polyps. The gastrointestinal adenomas predominantly arise in the colon and rectum, but small intestinal and gastric lesions also develop.
Turcot syndrome is linked to the APC locus. Hamilton et al detected genetic abnormalities in 13 of 14 registry families (75). Germline APC mutations were detected in ten. The glioblastomas and colorectal tumors in three of the families and in the original family studied by Turcot also had replication errors characteristic of hereditary nonpolyposis colorectal cancer (HNPCC). In addition, germline mutations in mismatch repair genes MLH1 or PMS2 were found in two families. Thus, the association between the brain tumors and multiple colorectal adenomas can result from two distinct types of germline defects (i.e., mutation of APC or a mismatch repair gene) (75).
TABLE 12.4 Lewis Classifications of Turcot Patients | ||||||
|---|---|---|---|---|---|---|
|
MYH Adenomatous Polyposis
In 2002, Al-Tassan et al reported a Welsh family in which three members had multiple colorectal adenomas and carcinoma with an autosomal recessive pattern of inheritance (78). Their tumors demonstrated an excess of somatic mutations in APC, all of which represented G:C to T:A substitutions. The authors observed that such mutations are frequently the result of oxidative injury to DNA and, therefore, tested oxidative repair genes for germline changes in these patients. They found that affected patients carried two missense mutations (Y165C and G382D) in the human base excision repair gene MYH, located on chromosome 1. Subsequent studies by others have confirmed the association between multiple adenomatous polyps, early-onset adenocarcinoma, and biallelic germline mutations in MYH (79,80). Besides the two originally described MYH mutations, other mutations have now been observed including truncating, missense, in-frame insertions and putative splice site mutations (78,80,81).
The MYH protein functions as a base excision repair DNA glycosylase that excises adenines incorrectly incorporated opposite 8-oxo-7,8-dihydro-2’-deoxyguanosine, one of the most stable products of oxidative DNA damage (82). Adenomas and carcinomas from patients with inherited defects in MYH demonstrate an excess of G:C to T:A transversions in both the APC and ras genes (78,83).
Germline abnormalities in both MYH alleles correlate with the number of polyps observed clinically. The likelihood of MYH mutations increases with increasing numbers of polyps (80). However, bilallelic germline MYH mutations may occur in patients with early-onset colorectal carcinoma without polyposis (81,84). Some data suggest that heterozygous MYH mutations may also associate with an increased risk for carcinoma, and may act in an autosomal dominant mode of inheritance with relatively low penetrance (84). In fact, 47% of colorectal carcinomas arising in patients with monoallelic MYH mutation show loss of heterozygosity at the MYH locus, a finding that suggests that MYH may be an important factor in cancer development in this group of patients. In addition, patients with monoallelic MYH mutation more commonly report a family history of colorectal
cancer (84). Loss of heterozygosity at MYH has been observed in sporadic colon carcinomas (85).
cancer (84). Loss of heterozygosity at MYH has been observed in sporadic colon carcinomas (85).
Nonhereditary Adenomatous Polyposis Syndromes
Multiple Colonic Adenomas
The presence of multiple adenomas (<100 in the colorectum) defines a group of patients without a clear genetic disorder, but who exhibit an increased risk for developing colon carcinoma. Morson surveyed patients with intestinal neoplasia at St. Mark’s Hospital and found 1,846 individuals who had multiple adenomas (86). Of these, 27.9% had more than one adenoma; 4.5% had more than five adenomas. In this series, the patients with familial polyposis had a minimum of 200 polyps, whereas the nonfamilial group had a maximum of 48. The percentage of patients with associated carcinoma increased with increasing numbers of adenomas; 80% of patients with 6 to 48 polyps had a carcinoma (86). Many of these patients may in actuality have AAPC or MYH polyposis.
Hyperplastic Polyposis
Hyperplastic polyposis (HP) is a rare syndrome first described in 1980 as “metaplastic polyposis” (87). In the original series, seven patients were described, each with more than 50 colonic hyperplastic polyps. None of the patients in the group developed colonic adenocarcinoma. However, numerous reports have subsequently described the occurrence of colorectal cancers in HP patients (88,89,90), suggesting that patients with this syndrome are at increased risk for colon cancer development. Familial aggregation of the disorder has been observed in some patients, but a definite genetic association has not been demonstrated (91).
Patients with hyperplastic polyposis develop not only hyperplastic polyps, but also sessile serrated polyps, serrated adenomas, classic adenomas, and mixed serrated and adenomatous polyps (Fig. 12.15). The World Health Organization (WHO) criteria for diagnosis of the syndrome are summarized in Table 12.5 (92). The mean age at diagnosis is 52 to 61 years (90), although the disease has also been reported in younger patients. Both sexes appear to be affected equally. Most patients have more than 30 but fewer than 100 polyps.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


