Pediatric Renal Tumors and Neuroblastoma
Satish K. Tickoo
RENAL TUMORS
Renal tumors in children are uncommon and constitute a clinicopathologic spectrum quite different from that in the adults. Each year, approximately 500 new cases are diagnosed in the United States. These relatively small numbers make it very difficult for most pathologists to be familiar with their proper diagnosis. Staging for renal tumors is based on National Wilms Tumor Study Group (NWTSG) and its successor, the Children’s Oncology Group (COG) staging system, and International Society of Paediatric Oncology (SIOP) system, particularly in Europe (1,2,3,4). In both the COG and SIOP protocols, biopsies of the renal tumors are not recommended, and tumors are upstaged to stage III if any biopsies are performed on them. This is based on the assumption that preresection biopsies are highly likely to lead to tumor spillover into the abdominal cavity. However, studies by United Kingdom Children’s Cancer Study Group (UKCCSG) in which needle core biopsies were performed preoperatively on suspected Wilms tumors indicate that percutaneous biopsy of such masses is relatively safe and effective (5). Management of the tumors under COG and SIOP protocols differ significantly. The COG approach relies on primary nephrectomy followed by further therapy based on the pathologic evaluation of the specimen (1), whereas the SIOP protocol advocates preoperative chemotherapy first, followed by surgery, and further therapy based on stage and histology, and risk stratification of the residual mass (3). The diagnosis in SIOP protocols is primarily based on characteristic imaging features of the tumor. On the other hand, UKCCSG in the United Kingdom recommends a needle core biopsy for the initial diagnosis, followed by chemotherapy and subsequent SIOP-based management (5). Thus, performance of needle core biopsies of renal tumors in children, in general, is very infrequent. However, biopsies may be performed in masses for diagnosis of tumors in patients with advanced disease before instituting preoperative chemotherapy, and for documentation of metastases/recurrence or contralateral disease (6), even under COG protocols.
Although Wilms tumor constitutes an overwhelming majority of renal tumors in pediatric age group, its differentiation from other tumor types is an absolute necessity, as chemotherapeutic options are significantly different among different tumor types.
Although Wilms tumor constitutes an overwhelming majority of renal tumors in pediatric age group, its differentiation from other tumor types is an absolute necessity, as chemotherapeutic options are significantly different among different tumor types.
WILMS TUMOR/NEPHROBLASTOMA
Wilms tumor constitutes approximately 87% of all renal masses in the pediatric age group (7,8). The peak incidence is between 2 and 3 years of age, with 98% of cases occurring in children younger than 10 years of age. On radiologic imaging, most of these appear as well-circumscribed solid tumors with a pseudocapsule, distorting the renal parenchyma and collecting system (9). Only the pure blastemal or blastema predominant type of the tumor may have an infiltrative margin. Thus, imaging allows for a presumptive diagnosis of Wilms tumor in most cases.
Wilms tumor typically shows a triphasic morphology—blastemal, epithelial, and stromal (Fig. 12.1A)—many may show preponderance (Fig. 12.1B) or exclusive presence of only one component. Blastemal component is composed of closely packed, small blue round cells with scant cytoplasm, evenly distributed coarse chromatin, and no, or at the most small, nucleoli. Prominent mitoses, apoptotic figures, and nuclear overlap are characteristic (Fig. 12.2A). Epithelial component shows variable differentiation toward tubules (Fig. 12.2B) or glomeruli (Fig. 12.2C) and can range
from primitive rosette-like tubules to well-formed maturing and mature tubules, ill-formed glomerular structures, and variable papillary architecture (Fig. 12.2D). Occasionally, mucinous or squamous differentiation may also be present. Tumors with extensive heterologous differentiation are designated “teratoid Wilms tumor” by some. Mesenchymal component usually consists of nondescript spindle cells (Fig. 12.1A), but smooth muscle,
skeletal muscle (Fig. 12.2E), or fibroblastic differentiation is not infrequent. Occasionally, fat, cartilage, bone, ganglion cells, or neuroglia may be present.
from primitive rosette-like tubules to well-formed maturing and mature tubules, ill-formed glomerular structures, and variable papillary architecture (Fig. 12.2D). Occasionally, mucinous or squamous differentiation may also be present. Tumors with extensive heterologous differentiation are designated “teratoid Wilms tumor” by some. Mesenchymal component usually consists of nondescript spindle cells (Fig. 12.1A), but smooth muscle,
skeletal muscle (Fig. 12.2E), or fibroblastic differentiation is not infrequent. Occasionally, fat, cartilage, bone, ganglion cells, or neuroglia may be present.
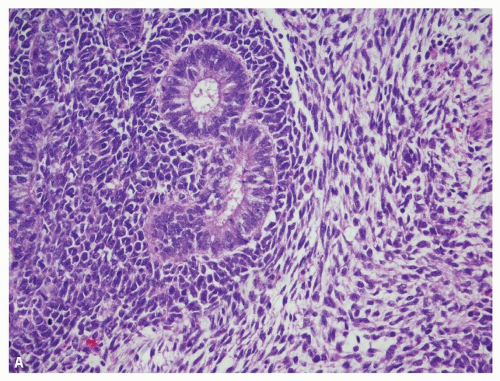 FIGURE 12.1 Wilms tumor/nephroblastoma. A: A tumor with typical triphasic histology showing the blastemal, epithelial, and stromal components. B: Another tumor containing only the blastemal and epithelial components (biphasic). |
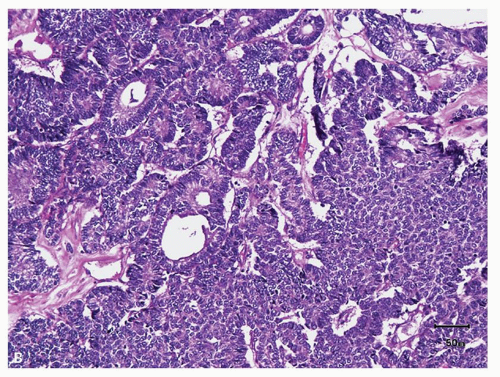 FIGURE 12.1 (Continued) |
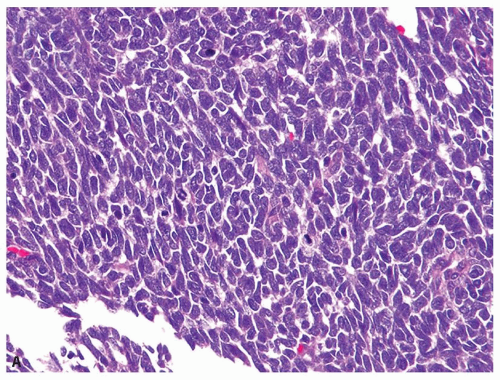 FIGURE 12.2 Wilms tumor. A: Typical histology of blastema. B: Epithelial component composed of primitive tubules. C: Better differentiated tubules and attempted glomerular formation (arrow). D: Papillary architecture in the epithelial component. E: Rhabdomyomatous differentiation in the stromal component. |
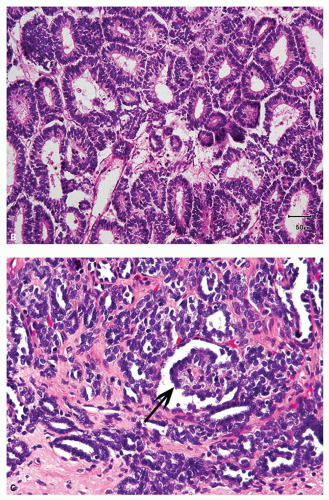 FIGURE 12.2 (Continued) |
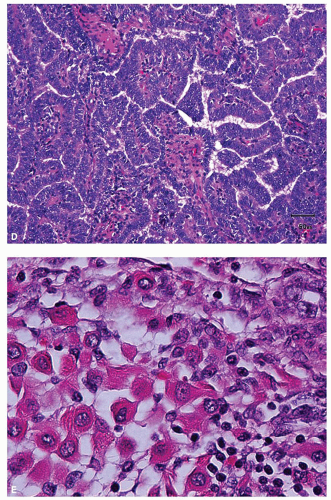 FIGURE 12.2 (Continued) |
One of the most important pathologic contributions influencing the management of Wilms tumor in the resection specimens is the determination of “favorable” or “unfavorable” histology (1,4). Approximately 5% of Wilms tumors show diffuse anaplasia in the resection specimens. Features of anaplasia include markedly enlarged tumor cell nuclei with hyperchromasia and multipolar mitotic figures (Fig. 12.3). Anaplasia is rare before 2 years of age but involves 13% of tumors beyond age 5 years (10). Anaplasia correlates with resistance to chemotherapy and with p53 mutations in the tumor (1,3,4). However, only diffuse anaplasia is clinically/therapeutically important. Its differentiation from focal anaplasia is essential. Because of the diagnostic complexities associated with determination of diffuse versus focal anaplasia, as well as the limitation of only limited amount of tumor being sampled, needle biopsies may not be particularly useful in this determination (5,6). However, it is essential that the presence of anaplasia be reported when observed in needle core biopsies. Reporting of anaplasia is also important in posttherapy specimens. Posttherapy tumors with anaplasia or blastemal type histology are both regarded as high-risk tumors in SIOP protocols and are consequently managed with more aggressive subsequent therapies (3).
Ancillary Studies
Immunohistochemical support may be needed particularly on needle core biopsies, mainly in tumors composed of blastemal elements alone, but
sometimes also in others (11). WT1 often, although not always, shows immunoreactivity usually limited to blastemal and epithelial elements; stromal components are mostly negative (Fig. 12.4). Blastemal cells may sometimes label for desmin, but other muscle markers such as actin, myogenin, and myoD1 are negative (12). Cytokeratin is usually negative or at the most focally positive in blastema but is usually positive in epithelial components. Recently, the stem cell marker SALL4 was also reported to be expressed in the tumor (13).
sometimes also in others (11). WT1 often, although not always, shows immunoreactivity usually limited to blastemal and epithelial elements; stromal components are mostly negative (Fig. 12.4). Blastemal cells may sometimes label for desmin, but other muscle markers such as actin, myogenin, and myoD1 are negative (12). Cytokeratin is usually negative or at the most focally positive in blastema but is usually positive in epithelial components. Recently, the stem cell marker SALL4 was also reported to be expressed in the tumor (13).
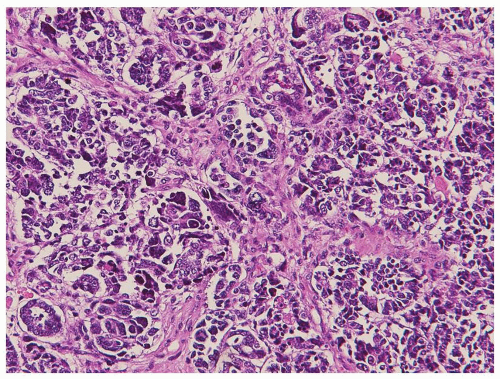 FIGURE 12.3 Wilms tumor with anaplasia. |
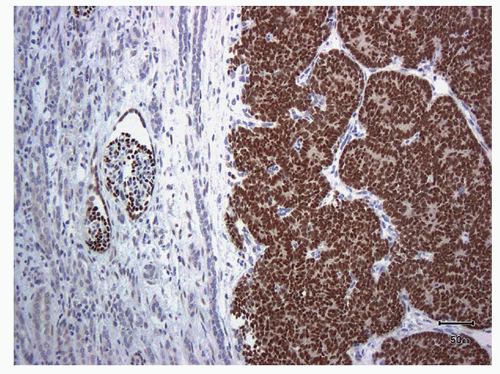 FIGURE 12.4 Immunohistochemical stain for WT1 staining the blastemal and epithelial components, but not the stroma. Positivity in glomeruli acts as internal positive control. |
Differential Diagnosis
Blastemal Wilms tumor requires differentiation of from other small blue round cell tumors, including neuroblastoma, rhabdomyosarcoma, and primitive neuroectodermal tumor (PNET). The presence of nuclear molding, early tubular differentiation with organized nuclear alignment around early lumina is typical of nephroblastoma, and the presence of any true tubular lumina always favors Wilms tumor. Presence of the fibrillary neuropil always supports a neuroblastoma. Immunohistochemical stains may be required in small biopsies to separate Wilms tumor from these other possibilities (neuroblastoma, synaptophysin/chromogranin+, WT1-; PNET, CD99+, WT1-; rhabdomyosarcoma, myogenin/myoD1/desmin+, WT1-).
Epithelial-predominant Wilms tumor needs to be differentiated from metanephric adenoma, and rarely solid and predominantly tubular, type 1 papillary renal cell carcinoma (RCC). Metanephric adenoma shows uniform, nonoverlapping nuclei with delicate chromatin and inconspicuous
nucleoli and marked paucity of mitotic figures. Immunostain for WT1 may be negative or weak and focally positive in metanephric adenoma compared to usually strong and diffuse in Wilms tumor. Papillary RCC, type 1, often shows foamy macrophages. Nuclei do not appear primitive and AMACR and CK7 are diffusely and strongly positive in type 1 papillary RCC, whereas WT1 is usually negative. In Wilms tumor, AMACR is negative, CK7 is usually negative or at the most only focally positive, and WT1 shows diffuse and strong positivity.
nucleoli and marked paucity of mitotic figures. Immunostain for WT1 may be negative or weak and focally positive in metanephric adenoma compared to usually strong and diffuse in Wilms tumor. Papillary RCC, type 1, often shows foamy macrophages. Nuclei do not appear primitive and AMACR and CK7 are diffusely and strongly positive in type 1 papillary RCC, whereas WT1 is usually negative. In Wilms tumor, AMACR is negative, CK7 is usually negative or at the most only focally positive, and WT1 shows diffuse and strong positivity.
Cellular mesoblastic nephroma can be mistaken for Wilms tumor. WT1 does not stain mesoblastic nephroma. Unlike Wilms tumor, malignant rhabdoid tumor shows prominent nucleoli and cytoplasmic inclusions. While WT1 is negative in rhabdoid tumors, they also characteristically show lack of nuclear staining for BAF47 (INI1). Rarely, clear cell sarcoma may mimic Wilms tumor. However, clear cell sarcoma often shows a characteristic, branching “chiken-wire” like vasculature. The chromatin is relatively pale, and WT1 is negative.
Other important differential diagnostic possibilities for Wilms tumor are the hyperplastic or neoplastic nephrogenic cell rests. On limited material, such as a needle core biopsy, the distinction can be very difficult to be made based purely on cytoarchitecture. Presence of a pseudocapsule strongly favors Wilms tumor over a cell rest, as neither the hyperplastic nor the neoplastic cell rests show the presence of a well-formed pseudocapsule (14,15). Otherwise, the differentiation always needs clinical and radiologic support.
CLEAR CELL SARCOMA
Clear cell sarcoma of the kidney shows peak incidence between 2 and 3 years of age, although the range varies from 2 months to 14 years of age (16,17,18,19). Rare congenital cases have been reported, and occasional cases in adults have also been described (20).
Typical morphologic features are present at least focally in about 90% of the tumors and consist of an almost evenly distributed network of vascular septa, with branching chicken-wire pattern (Fig. 12.5A), somewhat akin to that seen in myxoid liposarcoma and oligodendroglioma (16). These septations divide tumor into nests and cords of cells. The polygonal tumor cells typically have indistinct cell borders, and the nuclei show finely granular chromatin and inconspicuous nucleoli (Fig. 12.5B). The cells are often surrounded by pale, mucopolysaccharide-rich material that often creates the illusion of clear cytoplasm (Fig. 12.5B). Necrosis may occasionally be present and is considered as the only independent histologic factor for adverse prognosis. Along the periphery of the tumor, entrapped native renal tubules are often observed.
Variant histologic patterns in the tumor are not very uncommon and are expected to create diagnostic difficulties on needle core biopsies as they often do in resection specimens. Such variant patterns may include
tumors with prominent myxoid stroma, or sclerosing, cellular, epithelioid trabecular, palisading (Verocay body-like), spindle cell, storiform, and anaplastic types. Although the histologic variants do not affect prognosis, they can make diagnosis very difficult on limited material (5). Clear cell sarcoma is one of the most common pediatric renal tumors that can be misdiagnosed on needle core biopsies (5).
tumors with prominent myxoid stroma, or sclerosing, cellular, epithelioid trabecular, palisading (Verocay body-like), spindle cell, storiform, and anaplastic types. Although the histologic variants do not affect prognosis, they can make diagnosis very difficult on limited material (5). Clear cell sarcoma is one of the most common pediatric renal tumors that can be misdiagnosed on needle core biopsies (5).
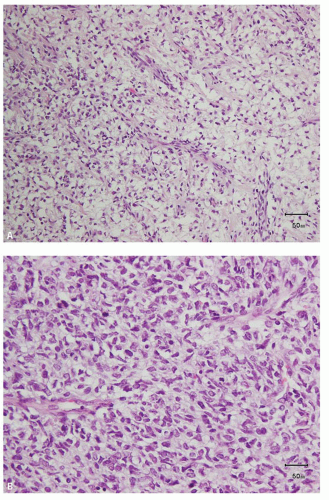 FIGURE 12.5 Clear cell sarcoma. A: The characteristic pale appearance and the typical vasculature. B: The vasculature is often retained even in the cellular areas. |
Ancillary Tests
The diagnosis is primarily based on histologic criteria, particularly the type of vasculature, as there are no specific immunohistochemical features for the tumor available at present (16). Immunohistochemical staining is mainly useful for excluding other renal tumors. The tumor cells are immunoreactive for vimentin and BCL-2. Stains for epithelial markers are almost always negative, as are WT1 and CD99. Immunoreactivity for p53 is consistently observed in tumors with anaplastic features only. Recurrent translocation between chromosomes 10 and 17 has been reported in a few cases and was recently found to be present in 12% of clear cell sarcomas of the kidney (21).
Differential Diagnosis
Unlike clear cell sarcoma, Wilms tumor shows coarse chromatin pattern, often with nuclear overlapping and molding. WT1 is also mostly positive in Wilms tumor. PNET can be differentiated by its positivity for CD99 and FLI-1. Cellular mesoblastic nephromas can be distinguished from clear cell sarcoma with the lack of the chicken-wire-type vascular. The distinction may also be helped by the consistent positivity for smooth muscle actin (SMA) in mesoblastic nephroma. Unlike clear cell sarcoma, malignant rhabdoid tumor almost always shows very prominent nucleoli and frequent presence of cytoplasmic large inclusion. The consistent lack of nuclear positivity for BAF47 (INI1) in malignant rhabdoid tumor is also a very useful diagnostic aid.
MALIGNANT RHABDOID TUMOR
Malignant rhabdoid tumor of the kidney (MRTK) comprises approximately 2% of pediatric renal tumors, presenting at a mean age of 1 year (22,23,24). Approximately 80% of the cases are reported before the age of 2 years; the tumors are virtually nonexistent after 5 years of age. Most of the cases present with stage III or IV disease; an overwhelming majority of stage IV renal tumors that present by 7 months of life are MRTK. Tumors similar to that in the kidney also exist in extrarenal sites, including central nervous system (atypical teratoid/rhabdoid tumor) and soft tissue. Germline mutations of INI1 are common in patients with apparent sporadic tumors as well as in familial rhabdoid tumor predisposition syndrome (25).
Histologically, the tumor shows sheets of monotonous loosely cohesive large, ovoid to polygonal cells, with high nuclear grade, extensively infiltrating the renal parenchyma (Fig. 12.6A). The characteristic cytologic features include vesicular chromatin, prominent eosinophilic nucleoli, and intracytoplasmic hyaline, pink inclusions (Fig. 12.6B). The inclusions may be sometimes difficult to find; looking for them around necrotic foci increases the likelihood of finding these inclusions. The tumor often shows brisk mitotic activity and areas of necrosis. Vascular invasion and
extrarenal extension are quite common. Variant histologic features, often present in association with typical areas, include sclerosis, spindling, pseudopapillary formation, and cystic change, among others (22,23,24).
extrarenal extension are quite common. Variant histologic features, often present in association with typical areas, include sclerosis, spindling, pseudopapillary formation, and cystic change, among others (22,23,24).
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


