Clinical Findings
Most infants with biliary atresia are term infants with normal birth weights. Infants present within the first 1 to 6 weeks of life with jaundice, pale stools, and dark urine. Other findings include pruritus and failure to thrive. Failure to thrive can manifest as poor weight gain or excessive feeding.2 Serum abnormalities included conjugated hyperbilirubinemia, high γ-glutamyltransferase (GGT) levels, high alkaline phosphatase levels, and elevated cholesterol but normal triglyceride levels.
Associated anomalies are present in about 20% cases. The most common (10% of total cases) is called the biliary atresia-splenic malformation syndrome and is associated with polysplenia, asplenia, or a double spleen.2 Other associated malformations with this syndrome include situs inversus, a preduodenal portal vein, intestinal malrotation, absent vena cava, cardiac anomalies, and pancreas anomalies.
Etiology
The etiology is unknown, although many possibilities have been proposed. An outbreak of biliary atresia in lambs was associated with toxin exposure,3 but currently, no specific toxin has been associated with human cases.
Treatment
Treatment currently is focused on surgical intervention called a Kasai procedure, or hepatoportoenterostomy, where the remnants of the extrahepatic tree are removed and a loop of the duodenum is attached to the hilum of the liver. The Kasai procedure is not curative and is used as a bridge to liver transplantation. The best outcomes after the Kasai procedure are observed in younger individuals, usually defined as those less than 60 days after birth. There is some data that indicates that infants undergoing the Kasai procedure can further benefit from corticosteroid therapy, antibiotic therapy, or bile flow stimulating agents, all of which help to improve bile flow, although their long-term benefit remains unproven.4
Imaging Findings
Ultrasound typically shows an enlarged liver with no dilatation of the common bile duct. The gallbladder is commonly absent or contracted but can be present and appear relatively normal in subset of cases. A hepatobiliary iminodiacetic acid (HIDA) scan typically shows good hepatic uptake but reduced or absent intestinal excretion within 24 hours. However, reduced or absent intestinal excretion on HIDA scans is not specific for biliary atresia and can be seen in other pediatric cholestatic liver diseases. Currently, there is only a limited diagnostic role for endoscopic retrograde cholangiopancreatography (ERCP) or magnetic resonance retrograde cholangiopancreatography (MRCP) because of technical limitations, but this may change in the future.
Histologic Findings
The histologic findings in biliary atresia depend on when the biopsy is performed in the course of the disease. The earliest biopsies (days after birth) show the least specific findings, with variable cholestasis, extramedullary hematopoiesis (EMH), and little, if any, bile ductular proliferation. Fortunately, most biopsies are not obtained in this early phase of the disease.
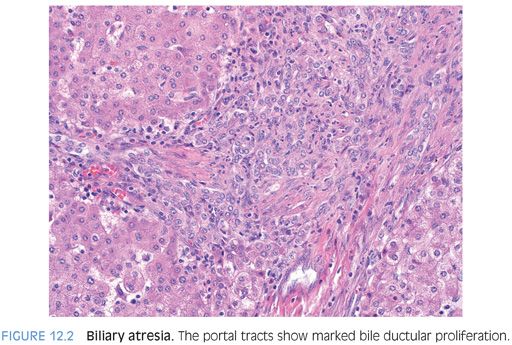
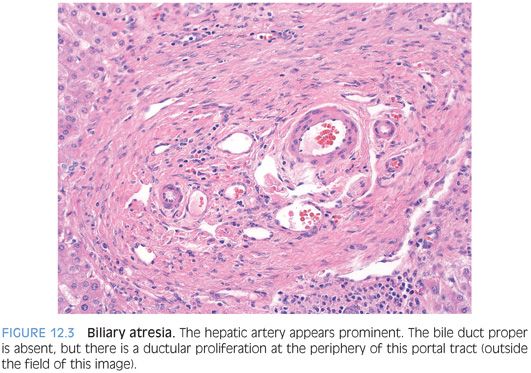
PORTAL TRACTS. Bile ductular proliferation is the major finding in most cases (Fig. 12.2), often accompanied by a prominent hepatic artery (Fig. 12.3) and some degree of portal fibrosis (eFig. 12.1). The amount of portal fibrosis will vary depending on the time of the biopsy. The hepatic artery often appears prominent because of thickening of the muscular wall. This finding is not specific for biliary atresia, however, because it can be seen in a number of different cholestatic liver diseases. The bile ductular proliferation will vary in intensity depending on the time of the biopsy. Liver biopsies obtained early in the disease course can have very mild changes. In most biopsies, however, the ductular proliferation is a dominant finding. In some cases, bile plugs can be seen in the proliferating ductules.
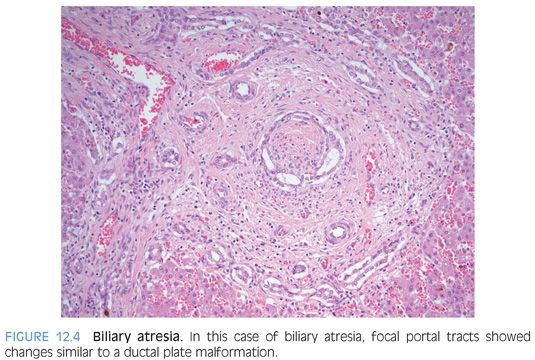
In a subset of approximately 20% to 50% of cases,5–7 the ductular reaction can have a pattern that suggests a ductular plate malformation (Fig. 12.4), with proliferating ductules forming a circular structure that resembles the early ducts in liver organogenesis. The significance of this finding is unclear. Some authors have suggested it represents an intrauterine start to the biliary atresia,8 whereas others have suggested a worse prognosis.6,9 However, the prognostic finding has not been validated by others.7,10 These biliary changes that resemble ductal plate malformation are not associated with the biliary atresia-splenic malformation syndrome.11
The hepatic lobules show cholestasis with relatively little to no inflammatory changes. The hepatocytes can also show giant cell transformation. EMH is common.
The amount of fibrosis will vary depending on the time of the biopsy. Earliest biopsies can show little or no fibrosis, but in most cases, there will be at least mild portal fibrosis. Bridging fibrosis can also be seen and imparts a worse prognosis.
HILAR PLATE. A small portion of liver hilum is removed during the Kasai procedure. Histologic evaluation typically shows several small-sized bile ducts, occasional peribiliary glands, and lymphocytic inflammation, embedded in a background of fibrous and connective tissue.
IMMUNOSTAINS. The proliferating bile ductules seen on the liver biopsies in most cases of biliary atresia are CD56-positive (eFig. 12.2), whereas CD56-positive bile ducts are only rarely observed in other causes of pediatric cholestatic liver diseases.12–15 Immunostains for CD56 can be helpful but are best used in conjunction with the hematoxylin and eosin (H&E) findings. To date, no other immunostains have been reported to be useful.
PROGNOSTIC INFORMATION. The histologic findings in the liver biopsy as well as evaluation of the hilar plate removed during the Kasai procedure have both been reported to have prognostic information. Fundamentally, they all reflect the severity of the disease. Liver biopsies with more ductular proliferation and more fibrosis tend to do worse. Hilar plate sections with large-sized bile ducts (less atretic) tend to be better.
DIFFERENTIAL. Choledochal cysts and type 1 biliary atresia can appear similar on imaging studies, and liver biopsies can be helpful in making a definitive diagnosis. Although the liver biopsy findings in choledochal cysts can show ductular proliferation, the proliferation is typically milder and fibrosis is absent or mild. Bridging or worse fibrosis favors biliary atresia.16 In type 1 biliary atresia, the bile ducts and ductules are also typically CD56-positive.13
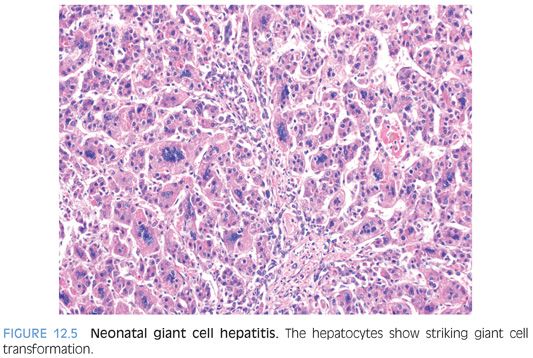
The histologic differential for biliary atresia often includes neonatal giant cell hepatitis (NGCH). In both diseases, giant cell transformation can be present and can sometimes be striking. Moderate or marked ductular proliferation, as well as portal fibrosis, all favors biliary atresia over NGCH. However, about 25% of cases of NGCH will have a mild ductular proliferation,17 and in some of these cases, the histologic findings will show substantial overlap with the early changes of biliary atresia. In fact, about 10% of biopsies initially diagnosed as NGCH will eventually be diagnosed as biliary atresia.17 Nonetheless, a diagnosis of biliary atresia is possible in most cases. Ductular proliferation, bile plugs in the ducts or proliferating ductules, and significant portal fibrosis, all favor biliary atresia.5
NEONATAL GIANT CELL HEPATITIS
Definition
NGCH is an important cause of cholestasis in infants. NGCH is a histologic pattern of injury that can only be diagnosed on biopsy. The biopsy shows giant cell transformation of the hepatocytes as the predominant finding, without evidence for biliary atresia or for paucity of intrahepatic bile ducts.
Clinical Findings
Clinically, infants with NGCH demonstrate elevated levels of conjugated bilirubin and have normal biliary trees by imaging studies. The clinical picture can often be confusing and liver biopsies are an important part of the diagnostic algorithm.
Etiology
NGCH is a pattern of injury that can be associated with a wide variety of injuries, including infections and genetic diseases (Table 12.1). A cause can be found in approximately half of all cases, but the remainder are idiopathic despite full clinical and histologic evaluation. Of the known etiologic associations, the most common is pituitary abnormalities with lack of normal hormone production.17

Histologic Findings
As an overview, NGCH is defined by syncytial giant cell transformation of the hepatocytes, with variable but generally mild inflammation and mild to moderate lobular cholestasis. In addition to these positive findings, the biopsies lack the marked obstructive changes of biliary atresia or the reduction in bile duct numbers that is typical of the various syndromes leading to paucity of intrahepatic bile ducts.
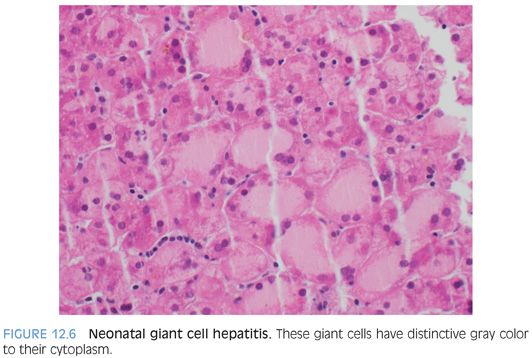
LOBULAR FINDINGS. Giant cell change is typically a striking finding at low power. Overall, about 40% of the hepatocytes are affected, although the percentage can range from as low as 5% to as high as 90%. In most cases, the hepatocytes with giant cell transformation will have either a predominately zone 3 distribution (one-third of cases) or have an azonal pattern, with no discernible zonation (two-thirds of cases). A zone 1 predominate pattern is only rarely observed. Hepatocytes with giant cell change will have between 4 and 10 nuclei in most cases (Fig. 12.5), although examples with up to 20 nuclei can occasionally be found. Giant hepatocytes often have abundant amphophilic cytoplasm that resembles endoplasmic reticulum proliferation with a peripheral localization of nuclei (Fig. 12.6). Other giant hepatocytes will have a homogenous eosinophilic cytoplasm with centrally located nuclei (eFig. 12.3). Both forms can be found in a single biopsy, although there often is a relative prominence of one of the morphologies.
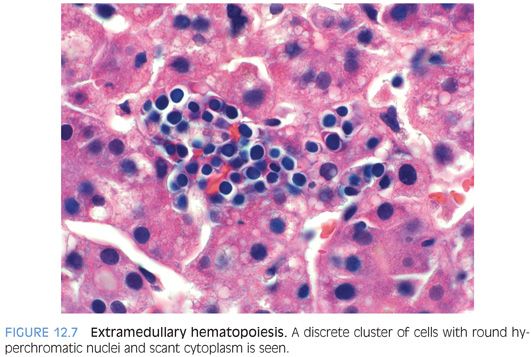
Lobular EMH is present in approximately three-fourths of cases and includes both myelopoiesis and erythropoiesis (Fig. 12.7). Lobular lymphocytic inflammation is mild or absent in nearly all cases. Lobular cholestasis is present in most cases and commonly shows moderate canalicular and mild to moderate hepatocellular cholestasis (eFig. 12.4). A small subset of cases will also show fatty change.17,18
PORTAL TRACT CHANGES. The portal tracts show mild to moderate cellularity, often with active myelopoiesis including numerous eosinophils and metamyelocytes. Portal lymphocytic inflammation is absent or mild in 95% of cases, with the remaining showing moderate levels of lymphocytic inflammation.
Bile ductular proliferation will be present on H&E stains in about 25% of cases and is usually mild. In addition, bile duct/ductular cholestasis can be rarely seen. Septal-sized bile ducts are only rarely sampled but can demonstrate mild epithelial lymphocytosis and equivocal epithelial injury. Small-sized bile ducts can appear hypoplastic and be difficult to identify on H&E stain,17,19 but cytokeratin immunostains can be used to rule out true ductopenia.
FIBROSIS AND IRON ACCUMULATION. Fibrosis is present in about one-third of cases. Fibrosis is usually mild, but a subset of cases can show bridging fibrosis. Livers with bridging fibrosis will often also show marked pericellular fibrosis. Isolated mild pericellular fibrosis can also be seen. A small series of cases have been published where reversed blood flow in the portal vein was associated with rapid fibrosis progression.20
Iron stains are negative in one-third of cases and in most of the remaining will show mild iron in hepatocytes, Kupffer cells, or both. In a subset of about 5% of cases, there can be moderate hepatocellular iron accumulation.
PAUCITY OF INTRAHEPATIC BILE DUCTS
Definition
Paucity of intrahepatic bile ducts is a pathology diagnosis but not a distinctive entity. Similar to NGCH, paucity of intrahepatic bile ducts represents a pattern of injury that can be associated with many different causes (Table 12.2).

Clinical Findings
The clinical presentation is typically an infant who presents with jaundice and conjugated bilirubin elevations in the first weeks to month of life. Neonates can have acholic stools, and HIDA scans can fail to secrete, mimicking biliary atresia.
Alagille syndrome is a distinct subset of paucity of intrahepatic bile ducts that is associated with JAG1 mutations and other congenital abnormalities including butterfly vertebrae, cardiovascular anatomic abnormalities, hypothyroidism, and pancreatic insufficiency.21
Histologic Findings
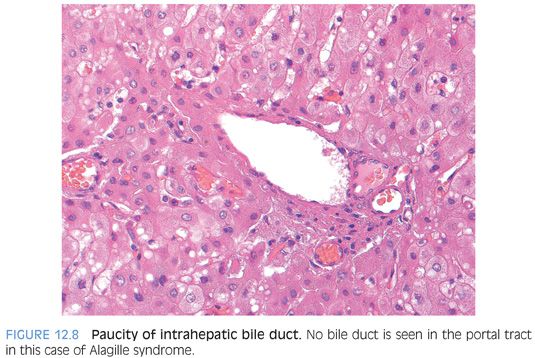
PORTAL TRACTS. The portal tracts show bile ducts that are hypoplastic or absent (Fig. 12.8). The portal tracts also show nonspecific inflammatory infiltrates that are predominately lymphocytic but can include eosinophils. The inflammation can range from mild to focally moderate and can sometimes obscure residual bile ducts. An immunostain for cytokeratin can help identify the bile duct loss. The portal tracts can also have mild bile ductular proliferation, but this should not be mistaken for the bile ducts proper (Fig. 12.9). In some cases, the ductular proliferation can have a distinctly immature phenotype, with the ductules also staining for hepatic markers (eFigs. 12.5 and 12.6). The bile duct loss initially affects the smaller branches of the biliary tree and can be patchy. Larger portal tracts, although uncommonly sampled, will have intact bile ducts (eFig. 12.7). Bile duct loss may not develop in some cases until after the first year or two of life. In one large series, 25% of liver biopsies did not have ductopenia in individuals who were older than a year in age and met clinical criteria for Alagille syndrome.22