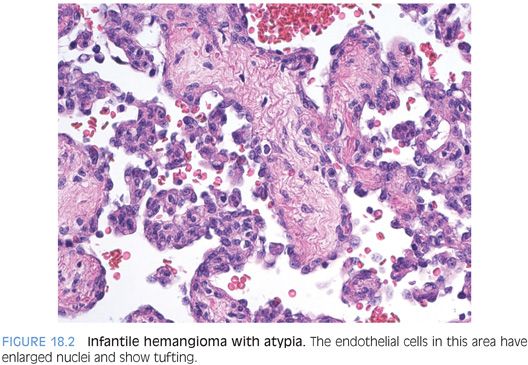
Nonetheless, infantile hemangiomas do have a risk of developing malignancy and should be examined for areas of atypia. Architectural atypia can include papillary tufts (Fig. 18.2) as well as solid areas (eFig. 18.1). In addition to atypia and endothelial tufting, other features suggesting angiosarcoma degeneration are spindly, kaposiform-like areas. Although rare, accumulated case reports and small series demonstrate a low but clear risk for aggressive behavior in some infantile hemangiomas. Currently, cytologic and/or architectural atypia appears to the best available histologic marker for aggressive potential. Although the clinical outcome in most cases is similar regardless of the presence or absence of atypia, almost all tumors with aggressive behavior have histologically atypical areas. Tumor recurrence is more common than metastases.
IMMUNOSTAINS. Infantile hemangiomas stain with vascular markers including CD31 and CD34. One study suggested GLUT1 may be helpful in separating infantile hemangiomas (GLUT1-positive) from vascular malformations (GLUT1-negative).1
MESENCHYMAL HAMARTOMA
Definition
A mesenchymal hamartoma is a benign mass lession of the liver composed of loose connective tissue, often with cystic change, admixed with benign bile ducts and occasional small islands or cords of hepatocytes.
Clinical Findings
Mesenchymal hamartomas usually present with nonspecific findings of an abdominal mass, but a wide range of clinical findings have also been reported. Serum AFP can be elevated in a subset of cases.4,5 There is a slight male predominance, and 85% of cases present before the age of 3 years.6 Mesenchymal hamartomas can also be detected by prenatal ultrasound. Rare cases have been reported in adults.7 Most mesenchymal hamartomas are single, but rare multifocal cases have been described.5
The etiology remains unclear, but chromosomal changes have been reported, frequently involving chromosome 19,8,9 suggesting that mesenchymal hamartomas are neoplastic in nature. Multiple case reports have also reported an association between mesenchymal hamartomas and placental mesenchymal dysplasia.10
Histologic Findings
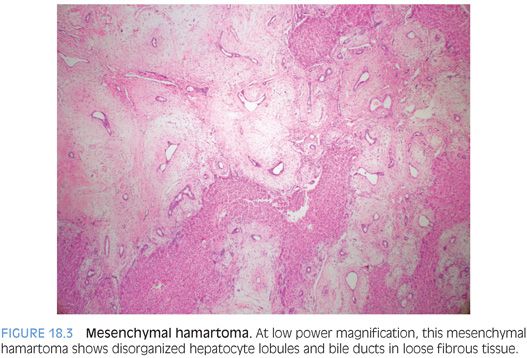
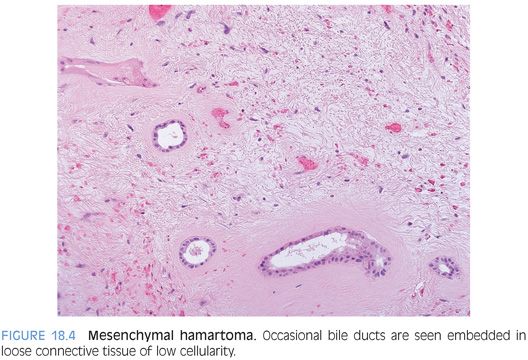
Mesenchymal hamartomas are composed of a variety of tissue types, but most commonly, the tumor is composed primarily of loose connective tissue with scattered benign bile ducts and occasional small hepatocyte islands (Fig. 18.3, eFig. 18.2). The connective tissue will show varying appearances, with some areas more densely collagenized and some areas more edematous or myxoid, but overall, the cellularity is low and there is no cytologic atypia (Fig. 18.4). The bile duct epithelium is also bland with no atypia and low mitotic activity. In some cases, the bile ducts will show a ductal plate malformation, whereas in others, the ducts will be dilated and form small cysts. The cysts can be microscopic in size or can be up to several centimeters. In some cases, the cystic areas can dominate the imaging or biopsy findings. In many of the larger cysts, no epithelial lining will be evident (eFig. 18.3) and such cysts may represent either degenerating cystic change of the mesenchymal tissue or biliary type cysts that have lost their epithelium. Small islands of hepatocytes can also be seen, especially at the periphery of the tumor. Also, a small vessel vascular proliferation can be seen at the periphery of some cases.6 The lesions generally show little or no inflammation, but extramedullary hematopoiesis is commonly found. Although mesenchymal hamartomas are benign, some cases can undergo malignant transformation to embryonal sarcoma.
Immunostains
The ductular epithelium lining the cysts and forming the bile duct structures is CK7 and CK19 positive. The loose connective tissue is typically vimentin-positive and can also be smooth muscle actin positive.11 The hepatocytes can be glypican 3 positive.12 In cases with elevated serum AFP, the hepatic islands and bile ducts within the hamartoma can be AFP-positive by immunostaining.4
EMBRYONAL SARCOMA
Definition
Embryonal sarcoma is an undifferentiated sarcoma most commonly seen in the pediatric population. In the literature, the term undifferentiated embryonal sarcoma or hepatic undifferentiated sarcoma is also used for this entity.
Clinical Findings
There is an equal male-to-female ratio, and the median age at presentation is around 10 years.13 However, embryonal sarcomas can also present in adults. The etiology is unknown, although a proportion arise out of mesenchymal hamartomas.14
Histologic Findings
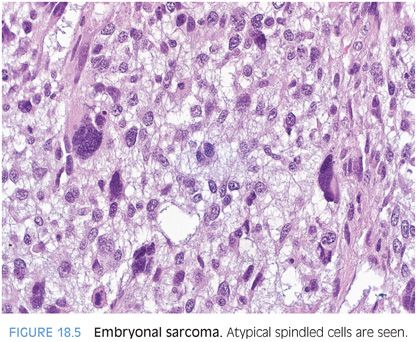
Most embryonal sarcomas are composed of undifferentiated spindled cells with significant and diffuse cytologic atypia (Fig. 18.5). The tumor cells tend to be medium to large in size and often have scattered areas of giant cell transformation (eFig. 18.4). The tumor cellularity can vary, with some areas becoming more fibrotic, but in many areas, the stroma is loose and myxoid in appearance. Hyaline globules can be seen in the tumor cells and sometimes outside the tumor cells. A periodic acid–Schiff (PAS) stain can be used to highlight the globules and they will also be resistant to diastase. Of note, biopsy specimens can sometimes lack hyaline globules due to sampling. Cystic degeneration can also occur and can dominate the radiologic and gross findings in some cases.15,16 Remnants of a mesenchymal hamartoma can be seen in some cases, in particular, in those arising in younger individuals.
Immunohistochemistry
There are a lot of case reports that describe immunostain findings but relatively few larger series. Nonetheless, overall, the tumors tend to show positivity for a variety of different cytokeratins including cytokeratin AE1/3 and CAM5.2.17–19 In most cases, the cytokeratin staining tends to be patchy. Some studies have reported a perinuclear dot-like positivity for cytokeratins AE1/3 and CAM5.2.17 Vimentin and CD68 are routinely positive.19,20 The tumor cells can also show membranous CD56 staining and focal CD10 membranous staining.17 Other stains that have been reported to be positive in a proportion of cases include α1-antitrypsin (positive in most cases) α1-antichymotrypsin, BCL-2, and P53.18,19 Desmin and α-smooth muscle actin show patchy staining in between 30% and 50% of cases.18,19,21 Tumor cells can be either diffusely or focally positive for glypican 3.12
Negative stains include myoglobin (focal positivity in rare cases), smooth muscle myosin, h-caldesmon, CD34 (focal positivity in rare cases), ALK1, S100 (focal positivity in rare cases), glial fibrillary acidic protein (GFAP), HMB-45, CD117, and hepatocyte paraffin 1 (Hep-Par1).18,19 Myogenin is negative.13,18
DIFFERENTIAL. In children, the primary differential is with biliary tract rhabdomyosarcoma. Biliary tract rhabdomyosarcomas tend to lack both the diffuse and striking anaplasia and the hyaline globules of embryonal sarcomas. Immunostains can also be helpful because myogenin and myogenic regulatory protein D1 (MyoD1) are typically negative in embryonal sarcomas but positive in most biliary tract rhabdomyosarcomas.13 In adults, metastatic sarcomas should be ruled out, including gastrointestinal stromal tumors.
OTHER RARE PEDIATRIC TUMORS
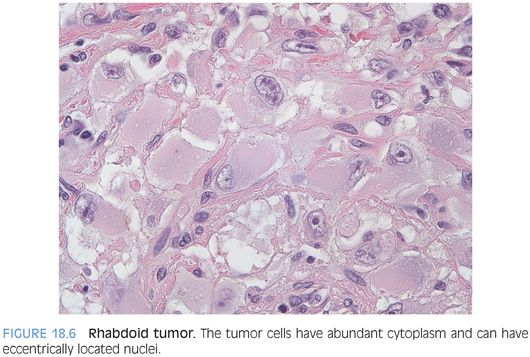
Rhabdoid Tumors
A recent review of the literature found that 34 rhabdoid tumors have been reported to date. The median age at presentation was 8 months, and about two-thirds of cases have metastatic disease at presentation.22 Cases have also been reported in adults.23,24 The clinical course is very aggressive with few long-term survivors.22 Because of the rarity of this tumor, clinical colleagues can easily confuse the term rhabdoid tumor with rhabdomyosarcoma and can sometimes benefit from a specific statement saying the tumor is not a rhabdomyosarcoma.
The tumors are composed of moderately sized epithelioid cells with abundant cytoplasm growing in sheets (Fig. 18.6, eFig. 18.5). In some areas, the tumor cells can have a spindled morphology. There is often significant necrosis, which can elicit a marked histiocytic infiltrate at the margins of the tumor. In some cases, the marked histiocytic inflammation can in part obscure the true diagnosis. The tumors uniformly show loss of nuclear INI1 immunostaining (eFig. 18.6). The immunostain profile is best studied in organs outside the liver,25 but the overall staining patterns are similar. Vimentin (eFig. 18.7) is positive in more than 90% of cases and smooth muscle actin in about 40% of cases, whereas pan-keratin staining is seen in 60% of cases (eFig. 18.8) and CAM5.2 in about 60% of cases (eFig. 18.9). Synaptophysin is positive in about two-thirds of cases, and S100 staining (often cytoplasmic) can be seen in a third of cases (eFig. 18.10). Other positive stains can include polyclonal carcinoembryonic antigen (CEA), CD34, and epidermal growth factor receptor (EGFR) (eFigs. 18.11 to 18.13). There is typically a high proliferative rate on Ki-67 immunostaining (eFig. 18.14). Immunostains are negative for chromogranin, CD34, HMB-45, desmin, myoglobin, and GFAP.
Angiosarcomas
Angiosarcomas in the pediatric population are very rare, and published data is sparse. They can show similar morphologic changes to adult angiosarcomas, but they can also have a kaposiform morphology, with solid areas of spindle cell growth containing small vascular slits. Focal “whorled areas” that resemble glomeruli have also been reported.26 The tumors are CD31- and CD34-positive.
Rhabdomyosarcoma
Usually, rhabdomyosarcoma (also called embryonal rhabdomyosarcoma) affects the extrahepatic bile ducts27 but has also been reported as an intrahepatic mass.28 The tumor cells can be spindled with eosinophilic inclusions or can be more rounded. Intermediate-shaped cells that resemble racquets can also be seen. Mitotic activity tends to be high, and there can be areas of necrosis and hemorrhage. The tumor can surround bile ducts leading to biliary obstruction. The tumor cells right underneath the bile ducts can appear more cellular, a finding referred to as a cambium layer. Cross-striations are hard to find, especially on a biopsy, and immunostains are helpful. Myogenin and MyoD1 are positive in most biliary tract rhabdomyosarcomas.13
BENIGN NODULAR LESIONS IN CHOLESTATIC PEDIATRIC LIVER DISEASE
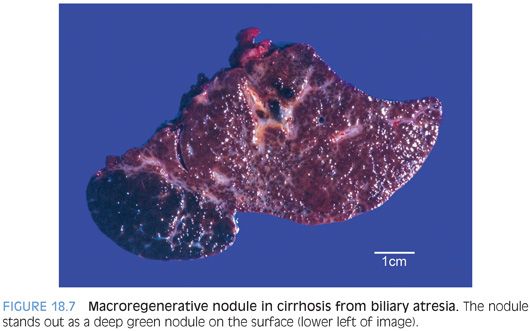
Approximately 5% of cirrhotic livers in the setting of cholestatic liver diseases due to biliary atresia can develop large nodules that on imaging studies can be worrisome for hepatocellular carcinoma.29 The nodules average 5 cm in size, but some can be larger than 10 cm. Almost all of these cases are macroregenerative nodules (Fig. 18.7) and not hepatocellular carcinoma. However, hepatocellular carcinomas also rarely occur in the setting of cirrhosis from biliary atresia (<1% of cases),30 so biopsies are often performed. The diagnosis of a macroregenerative nodule or hepatocellular carcinoma should be made using the same criteria as in adults.
FOCAL NODULAR HYPERPLASIA
Focal nodular hyperplasias develop in up to 8% of children who receive chemotherapy for various malignancies.31 They can occur many years after therapy, with a median interval of about 10 years.31 The frequency in the literature varies significantly, likely reflecting the length of follow-up as well as the chemotherapeutic agent. Some studies have suggested a link with high-dose alkylating agents or radiotherapy.32 The diagnosis of a focal nodular hyperplasia should be made using the same criteria as in adults. In the author’s experience, the most common difficulty in making this diagnosis in children is under appreciation that this lesion occurs in children.
PEDIATRIC HEPATIC ADENOMAS
Hepatic adenomas in children are rare but occur in several settings. The most common are glycogen storage diseases type I, type III, and less commonly, type VI.33–35 Rare cases of hepatic adenomas have also been reported in Hurler syndrome, severe combined immunodeficiency, and Fanconi anemia.36 Hepatic adenomas in the setting of Fanconi anemia are most commonly associated with androgen therapy and some can show significant cytologic atypia.36 The histologic diagnosis is made in the same manner as for adult hepatic adenomas. Hepatic adenomas in all of these conditions have a risk for malignant transformation.
HEPATOBLASTOMAS
Definition
Hepatoblastoma is a malignant epithelial tumor that shows varying degrees of hepatic differentiation and can also have a malignant sarcomatous component.
Clinical Findings
Epidemiology studies indicate that 91% of all pediatric liver tumors in those younger than 5 years of age are hepatoblastomas.37 The median age at presentation is approximately 18 months. Up to 5% may be present at birth, and approximately 70% are diagnosed before the age of 2 years. Hepatoblastomas do occur after the age of 5 years but are very rare and almost never seen past the age of 12 years. Hepatoblastomas may be increasing in frequency in the United States.37 There are several dozen case reports of hepatoblastomas occurring in adults,38 but many of these might possibly have better diagnoses, and overall, this group of cases likely represents a mixture of different tumor types, including many poorly differentiated, but otherwise ordinary, hepatocellular carcinomas with sarcomatoid features.
Risk factors are poorly understood, but there is a clear association with prematurity and low birth weight, especially with birth weight less than 1,500 g. A modest male predominance of 2:1 has been consistently identified. There is a large list of congenital anomalies that can co-occur with hepatoblastomas, but the frequency of any given finding is low. Overall, an estimated 15% of hepatoblastomas arise in the setting of known genetic syndromes.39 Of these, the strongest association to date has been with familial adenomatosis polyposis and the Beckwith-Wiedemann syndrome. The background livers typically show no histologic evidence of chronic liver disease and no fibrosis.
The clinical presentation is generally nonspecific but typically includes an enlarging abdomen and some degree of weight loss and anorexia. Many paraneoplastic syndromes have been described, but most of them are rare and do not provide unique insight into the diagnosis or etiology of hepatoblastomas.
Serum AFP levels are markedly elevated in >95% of cases and play an important role in patient workup and in monitoring response to tumor therapy. About 2% to 4% of cases will have normal or mild elevations (less than 100 ng/mL) in serum AFP at the time of diagnosis.40 Those hepatoblastomas that lack serum AFP elevations have a worse prognosis and typically have a small cell undifferentiated morphology or a mixed epithelial/mesenchymal morphology with rhabdoid features.
Many hepatoblastomas are treated in order to shrink the tumor prior to resection. Biopsies are often performed prior to the introduction of therapy. In most cases, the diagnosis is confidently made by needle or wedge biopsy, but cases composed of small undifferentiated cells can be challenging because the differential can include metastatic small round blue cell tumors such as Wilms tumor or neuroblastoma. In these cases, immunohistochemistry and electron microscopy can be very useful. Deeper levels can also be helpful because they can reveal areas with better differentiation that allow recognition of hepatic morphology.
Histologic Subtypes
Hepatoblastomas have been classified in different ways, but the most useful is that described in Armed Forces Institute of Pathology series and subsequently adopted by WHO (Table 18.1).










