Pancreatic Cancer: Molecular Biology and Genetics
Scott E. Kern
Eike Gallmeier
Michael Goggins
Ralph H. Hruban
Pancreatic neoplasia is a genetic disease. In all of its manifestations, pancreatic cancer is inextricably molded by a microevolutionary process involving the serial steps of genetic mutation and the consequent emergence of subclones having selective growth advantages.
The utility of this perspective has been borne out. Molecular genetic analyses of pancreatic neoplasms have led to the recognition of new histopathological variants of pancreatic cancer and to a better understanding of the cellular basis for differences in the clinical behavior of distinct types of neoplasms of the pancreas. For example, although most common ductal adenocarcinomas are relatively uniform clinically, pathologically, and genetically, there are less common variant forms that have differing histologic and genetic characteristics and that carry clinical distinctions of importance. Genetic assays can enable conspicuous groupings among the tumors (because a given neoplasm either has a mutation or it does not); therefore, this mode of analysis has spurred the recognition of these variant forms.
Ductal Neoplasia of the Pancreas
A variety of clinically and pathologically distinct neoplasms can involve the pancreas, and readers are encouraged to refer to the related chapters that review the histopathology (Chapter 27) and the epidemiology of pancreatic cancer (Chapter 24). Primary neoplasms of the pancreas are broadly divided into those having exocrine differentiation and those with endocrine differentiation. The infiltrating ductal adenocarcinoma is the most common malignant neoplasm having exocrine differentiation. The term ductal adenocarcinoma refers to a clinically aggressive neoplasm characterized by ductal differentiation, shown by its mucin production and the expression of certain cytokeratins. Ductal adenocarcinomas encompass the entire spectrum of histologic grades, from poorly differentiated to well differentiated, although the vast majority are moderately differentiated; the grades of differentiation per se have minimal clinical importance or demonstrable molecular associations. Among the ductal adenocarcinomas, however, there are some distinct molecular and histopathological variants (Fig. 25.1). In addition, there are a number of other exocrine neoplasms such as the intraductal papillary mucinous neoplasm (IPMN), which are clinically and pathologically distinct, although these variants are less well characterized at the molecular level.
Infiltrating ductal adenocarcinoma must also be distinguished from its precursor lesions, IPMNs, and pancreatic intraepithelial neoplasia (PanIN) (Fig. 25.2). PanINs are histologically distinct lesions in the pancreas, and molecular analyses of these distinct lesions provided hard evidence of the origins of and precursor lesions associated with infiltrating ductal adenocarcinoma (Fig. 25.2). IPMNs are usually larger than PanINs, although a unified classification independent of the size of the lesion may emerge on further work. IPMNs share some of the early genetic abnormalities of PanIN but lack some of the late stage genetic alterations, a difference that may help explain their more favorable tumorigenic course.
A “gatekeeper gene” analogous to the genes that prevent multiple adenomas in familial adenomatous polyposis in the colorectum (the APC gene) or endocrine hyperplasias in multiple endocrine neoplasia syndromes (e.g., the MEN1 gene) (1,2) has not been identified in the pancreas. Nonetheless, recent studies of pancreata surgically resected from individuals with a strong family history of pancreatic cancer reveal multiple foci of PanIN (3), suggesting that one day a gatekeeper gene may be discovered in the pancreas as well. Finally, it is important to note that, in molecular terms even more so than in histologic terms, ductal carcinomas of the pancreas are distinct from other intestinal neoplasias, such as the neighboring entity, the duodenal carcinoma. It is, however, more difficult to distinguish pancreatic ductal adenocarcinoma from the carcinomas arising at the duodenal papilla and from the distal common bile duct. Pancreatic adenocarcinoma can also be contrasted to another ductal neoplasm, breast carcinoma. There is a relatively homogeneous genetic profile for conventional ductal adenocarcinomas of the pancreas that largely mirrors the homogeneity in the histopathological appearance and in the aggressive clinical course. In breast carcinoma, this is not the case because a remarkable heterogeneity of molecular genetic patterns parallels its clinical and histologic heterogeneity.
Cytogenetics and Ploidy
In the analysis of global patterns of chromosome number and structure by classical cytogenetics, the karyotypes of short-term and established cultures of pancreatic adenocarcinoma reveal a very complex set of abnormalities, as is typical for most other carcinomas of adulthood. Translocations, whole chromosome and partial deletions, gains of chromosomes and segments thereof, and occasional minute chromosomal fragments are present (4,5). Despite a considerable heterogeneity in karyotypic patterns among the cells of individual pancreatic cancers (6), dominant patterns exist. Some of the recurrent patterns include deletions of chromosomal arms 9p, 17p, and 18q, sites of known major tumor-suppressor gene abnormalities. The CDKN2A/p16 tumor-suppressor gene resides on 9p, the TP53/p53 gene on 17p, and the SMAD4/DPC4 gene on 18q. The results of allelotyping, a molecular method of surveying the entire genome for chromosomal deletions (allelic losses), mirror these findings. The average allelotype indicates
that up to one-third of the chromosomal regions in ductal adenocarcinomas have suffered a loss of an allele and that the most common sites of loss involve chromosomal arms 9p, 17p, and 18q (7). Furthermore, there is a good correlation between the findings obtained with these two techniques, as a detailed comparison of cytogenetic abnormalities to allelotype data in a series of ductal adenocarcinomas demonstrated that most allelic losses are reflected in cytogenetic abnormalities (8).
that up to one-third of the chromosomal regions in ductal adenocarcinomas have suffered a loss of an allele and that the most common sites of loss involve chromosomal arms 9p, 17p, and 18q (7). Furthermore, there is a good correlation between the findings obtained with these two techniques, as a detailed comparison of cytogenetic abnormalities to allelotype data in a series of ductal adenocarcinomas demonstrated that most allelic losses are reflected in cytogenetic abnormalities (8).
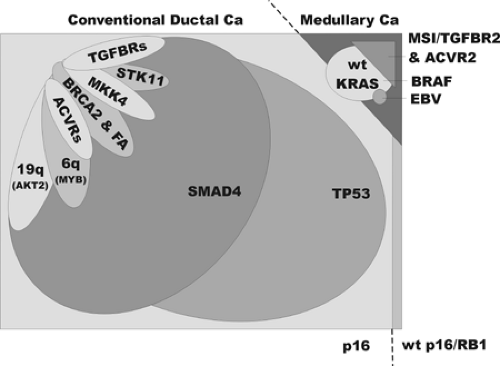 FIGURE 25.1. Venn diagram of the frequencies and relations of the gene mutations in pancreatic ductal adenocarcinoma. Each subset is labeled according to the histologic pattern or gene/chromosome abnormality and corresponds in area to the proportion of tumors having the genetic alteration (single alterations in the case of the KRAS oncogene, complex multiple alterations in the case of the amplifications of 19q and 6q, biallelic inactivation in the case of the tumor-suppressor genes). Medullary carcinomas encompass most cases having a wild-type KRAS gene and all cases having microsatellite instability (MSI). Most cases with MSI have a wild-type KRAS gene and a mutant BRAF gene, and all have had biallelic mutations of the polyadenine tract of the TGFBR2 and ACVR2 genes. Rare cases are reported to have mutation of the RB1 gene and might overlap with the small subset having an unmethylated wild-type p16 gene, reflecting a pattern of mutually exclusive gene mutation that is clearly defined in other tumor types. A single case harboring the Epstein-Barr virus episome is reported among the medullary cancers. The clustering of infrequently mutated genes in the upper left is meant to indicate the independent occurrence of these mutations that does not exclude their being present in the same tumors regardless of whether the tumors have alterations in TP53 or SMAD4; the particular subset that adjoins another and the degree of overlap has no intended meaning here. FA refers to the genes of the Fanconi anemia core complex. The constellation of tumor-suppressor mutations present in the medullary cancers and in those having the unmethylated wild-type p16 gene are currently poorly defined, and the diagram does not convey the relation of these tumors to this gene category. |
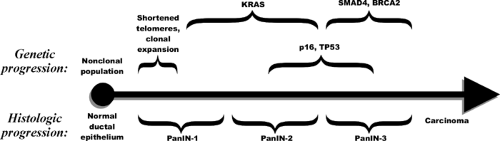 FIGURE 25.2. Genetic changes in the evolution of pancreatic ductal intraepithelial neoplasia (PanIN). PanIN is the new internationally defined system for classification of dysplasia (noninvasive epithelial neoplasia) of the pancreatic ducts. In brief, PanIN-1 comprises a flat mucosa of tall mucinous cells, PanIN-2 includes papillary architecture and some cytologic atypia, and PanIN-3 has a complex architecture with cytologic features resembling those of the invasive lesions, previously termed “carcinoma in situ.” KRAS mutations are not observed in the majority of PanIN-1 lesions, suggesting that there might be another, perhaps more universal alteration that initiates this neoplastic process. p53 Overexpression is best studied in PanIN-3 but occurs in at least some instances of PanIN-2. To date, PanIN-3 lesions are the only precursor stage in which the inactivation of both alleles of BRCA2 and SMAD4 been observed; thus, these genes appear to play a “late” role as mutational targets. |
The simple quantitation of total cellular DNA content by flow cytometry or static image cytometry can be used to compare the abnormal DNA content (termed aneuploidy) of cancer cells to normal cells, a ratio termed the DNA index (9). Measurements of DNA index have the advantage over classical cytogenetics in being applicable to the direct study of the primary tumor. However, these cytometric techniques can miss changes in individual chromosomes, and most tumors measured to have a normal DNA index do not in reality harbor a diploid chromosomal pattern (i.e., containing two copies of all 22 non–sex chromosomes).
There are many open questions regarding the cytogenetics of this pancreatic adenocarcinoma. For example, pancreatic cancer is similar to most carcinomas of adulthood in that a specific recurrent translocation has not been identified, although sensitive techniques to characterize the complex translocations of the carcinomas (in contrast to the simpler patterns seen in the hematopoietic malignancies) now exist. Spectral karyotyping is similar to karyotype analysis but is based on fluorescence in situ hybridization; it uses multiple fluorochromes, digital imaging, and computer analysis to represent a different color for each chromosome, allowing the recognition of constituent chromosomal parts involved in complex translocations in pancreatic cancers (10). Comparative genomic hybridization and related techniques such as the hybridization of tumor DNA to arrays of oligonucleotides, genes, and chromosomal segments have also been applied to pancreatic cancer (10,11); such techniques can be used to determine the relative copy number for each segment of chromosomal material and have proven useful for the identification of deletions and chromosome amplification (12). The most common sites of deletion and some notable sites of amplification are discussed later in this chapter. The findings of these techniques imply that pancreatic adenocarcinomas have considerable chromosomal instability, probably on the order of that seen in colorectal carcinoma.
Molecular Genetics
A simple molecular definition of a neoplasm is that it is a clone of cells distinguished from other tissues by autonomous growth and somatic mutations (13). These mutations lie within growth-controlling genes. Physiologically, neoplastic cells grow under what would otherwise be limiting conditions. Supporting and reactive tissues accompany the neoplastic cells in the formation of tumors. This definition clearly describes the infiltrating ductal adenocarcinomas of the pancreas and can be used to extend the concept of neoplasia to the precursor lesions (PanINs and IPMNs), where clonal genetic abnormalities in oncogenes and tumor-suppressor genes are readily demonstrable.
The molecular genetic alterations identified to date in infiltrating ductal adenocarcinomas of the pancreas now include instances of all five basic types of tumor mutation: translocation, amplification, deletion, subtle (intragenic) mutations, and the addition of exogenous (viral) sequences. As noted previously, only the translocations are not yet shown to form recurrent patterns that would indicate a specific selectable advantage, and the translocations observed might only represent the expected consequence of the general process that produces aneuploidy.
When a gene in a cancer is affected by mutation at a rate higher than would be expected by chance, we say that the gene is targeted by mutation. The mechanism increasing the prevalence of a mutation in a cancer is, of course, random, with selective pressures acting on the cellular population to favor the subclones harboring the mutated gene. The genes targeted by mutation can be grouped into three major classes. Oncogenes are genes whose function is activated by mutation (the normal gene is sometimes called a protooncogene). Oncogenes are said to be dominant because their appearance in a cell confers the subsequent cellular changes. Tumor-suppressor genes and genome-maintenance genes are inactivated by mutation. These are recessive gene classes in that the functional loss of both normal copies is needed to result in cellular changes, and the experimental transfer of a mutant version cannot be expected to confer a cellular change (an exception exists but represents yet another form of inactivated recessive gene: the dominant-negative mutation).
It is important to distinguish somatic mutations (those that occur during the life of the patient) from inherited (germline) mutations. Inherited mutations are transmitted from parent to child and will be present in all cells of the body. Inherited mutations usually, but not always, require a second (somatic) genetic event to manifest a cellular alteration. This second event, in the case of recessive genes such as the tumor-suppressor genes and genome-maintenance genes, is often the deletion of the remaining wild-type (normal) gene copy. In a line of analysis introduced by Nicholls and developed by Knudson, the neoplastic effects of an inherited mutation can be delayed because they are dependent on the chance occurrence of further somatic mutations (14,15).
Before considering a formal outline of molecular genetic events in pancreatic adenocarcinoma, it is appropriate to note a caveat of DNA methodology. Pancreatic carcinomas usually have a very characteristic and exuberant nonneoplastic host reaction, comprising dense scarring and chronic inflammatory reactions. For this reason, most cells in the mass formed by an invasive pancreatic ductal adenocarcinoma are genetically normal, reactive host cells such as fibroblasts and endothelial cells. Most genetic studies therefore use cell populations that are passaged in culture or in immunodeficient mice to enrich for neoplastic cells.
The following discussion presents the relationship of individual genetic changes to the major genetic and histologic classifications (Fig. 25.1), the means by which the genes were first discovered (Fig. 25.3), the frequencies of genetic changes, and major theories regarding their cancer-related functions (Fig. 25.4).
Oncogenes
The KRAS gene was first identified as the transforming gene of the Kirsten virus. Later, when DNA of human tumors was used to artificially transform mouse cells (i.e., conferring some of the properties associated with neoplastic cells), a mutated form of the ras genes was repeatedly isolated from multiple human tumor types (16). More than 90% of human pancreatic ductal adenocarcinomas have an activating point mutation in the KRAS gene (17). The occasional cases with wild-type KRAS genes tend to be the carcinomas with the medullary phenotype and BRAF gene mutations and include carcinomas with abnormalities in DNA mismatch repair (Fig. 25.1) (18,19,20). Most of the mutations in the KRAS gene result in codon 12 changing from glycine to aspartate or valine (although any possible mutation can be seen). In contrast, a broad spectrum of mutations is reported in colorectal carcinoma (21,22). The codon 12 KRAS gene mutations observed in most pancreatic cancers impair the GTPase function of the
ras protein; because the GTP-bound form of ras is the active form, this is believed to cause overactivity in various intracellular signals (23). Ras genes mediate the signals arising from the binding of growth factors, and major mitogen-activated protein kinase (MAPK) pathways are activated by K-ras activity (24).
ras protein; because the GTP-bound form of ras is the active form, this is believed to cause overactivity in various intracellular signals (23). Ras genes mediate the signals arising from the binding of growth factors, and major mitogen-activated protein kinase (MAPK) pathways are activated by K-ras activity (24).
 FIGURE 25.3. The history of discovery of genes targeted for mutation in pancreatic ductal adenocarcinoma. Most genes were first described from studies of general biology (e.g., kinase signal pathways or cell cycle mechanisms) or from artificial tumor systems (e.g., virally induced tumors in animals and in culture), and were only later shown to be mutationally involved in human tumors. Others were first identified by positional genomic approaches in human families and tumors. Most tumor-suppressor genes can be inactivated by a variety of genetic mechanisms. Slippage of simple repetitive DNA sequences is most characteristic of tumors with microsatellite instability at sites of extended repeats such as the polyadenine tract of the TGFBR2 and ACVR2 genes. Methylation of gene promoter sequences can affect various genes but is distinctly associated with complete transcript silencing for the p16 gene. There is a notable paucity of homozygous deletions among the mutational spectrum of the TP53 gene, an unusual finding among the tumor-suppressor genes. The restricted mutational spectrum shown here for the TGFBR1 and RB1 genes is likely only a manifestation of the low numbers of affected tumors reported to date. Mutational profiles are similar among the cancers of the pancreatic ducts and those of the intrapancreatic distal common bile duct. Ca, cancer. |
The BRAF gene is mutated in some medullary carcinomas of the pancreas (20). These same tumors have microsatellite instability and a wild-type KRAS gene. The BRAF gene stimulates the MAP kinase pathway, being positioned just downstream of ras genes.
Cyclin E is distinctly overexpressed in about 5% of pancreatic cancers, and, in one tumor subject to genetic analysis, this overexpression was attributed to a mutation of the FBXW7 (CDC4, a tumor suppressor) gene (20). Fbxw7 is an ubiquitin ligase responsible for the downregulation of the cyclin E protein. When mutated, cyclin E, an oncogene normally subject to cyclic expression during the cell division cycle, remains inappropriately expressed (25,26).
Gene amplification is seen in infiltrating ductal adenocarcinomas at a few chromosomal loci. These include the AKT2 gene and its neighboring genes on chromosomal arm 19q (10%–20% of cases), MYB and its neighbors on 6q (10% of cases), cyclin E, and others (10,20,27,28,29,30). It is assumed that the maintenance and expansion of these arrays of repetitive DNA is the result of selective pressures conferred by overexpression of the oncogenes contained within their boundaries. Unfortunately, the DNA segments involved (termed amplicons) are so large as to impair the clear identification of the true target gene(s) of the amplification process.
Tumor-Suppressor Genes
The tumor-suppressor genes are recessive genes and have a close association with inherited syndromes. The presence of one mutated copy in the germline is well tolerated during early development. The human gene pool thus includes mutated tumor-suppressor genes in at least 1% of some populations. Some inherited mutations are associated with the subsequent development of multiple early neoplasms as part of an obviously recognizable clinical syndrome, such as familial adenomatous polyposis or multiple endocrine neoplasia syndromes. The gene involved is termed a gatekeeper, in that its full inactivation of both copies is sufficient to initiate a lesion (2,13). As discussed previously, it has been suggested that the pancreas uses a gatekeeper form of proliferative control; however, this still remains to be proven. The evidence indicates that some inherited mutations of tumor-suppressor genes do not play an early role in neoplastic development for some defects appear to become biologically significant only later in tumorigenesis (31).
The tumor-suppressor genes targeted by mutation in pancreatic ductal carcinoma include the p16, TP53, SMAD4, TGFβ and activin receptors I and II, MKK4, STK11, and BRCA2 genes. These are discussed as follows. For these genes, intragenic sequence mutations act to inactivate the protein function and accompany loss of the wild-type gene copy. The intragenic mutations are of somatic origin, except where otherwise noted. All homozygous deletions noted are of somatic origin.
The p16 gene (CDKN2A, chr. 9p) was first identified from studies of inhibitors of the cell division cycle mechanism (32). It was later identified to lie at a known common site of homozygous deletions affecting chromosomal arm 9p in multiple tumor types and is the mutant gene that causes familial melanoma (33,34). The p16 gene may be the most commonly inactivated of the tumor-suppressor genes in humans. The p16 protein binds to the cyclin-dependent kinases Cdk4 and Cdk6 to inhibit their action. In the absence of p16, Cdk4 and Cdk6 phosphorylate and inactivate the cell cycle inhibitory protein Rb1 in late G1 phase, thereby initiating DNA replication. Because they lie within a single pathway, the p16 and RB1 genes have an inverse mutational relationship: when one is mutated in a particular tumor, the other is not (i.e., once p16 is mutated in a particular neoplasm, there should be no selective advantage to the further inactivation of RB1). The p16 gene is inactivated in virtually all pancreatic adenocarcinomas. Homozygous deletions or intragenic mutation are most common, although promoter methylation associated with the silencing of gene transcription occurs in the remainder (35,36). Rare mutations of RB1 are reported in pancreatic cancer (37).
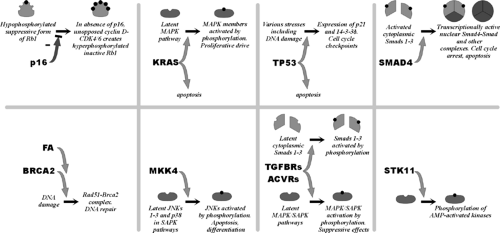 FIGURE 25.4. Pathways affected by mutation in pancreatic ductal adenocarcinoma. Major roles of each tumor-suppressor gene and the KRAS oncogene are indicated. Where the specific mechanism is known with some certainty, a straight arrow indicates the step mediated or facilitated, or in the case of p16, the step inhibited, by the wild-type gene. Mutations activate the function of the KRAS gene, whereas the other genes depicted are inactivated by mutation. Multiple additional properties are known for most of these genes; the discrimination of those functions with the highest relevance for tumorigenesis is an evolving subject of research and personal preference. Phosphorylation is depicted by occupation of a specific surface location of a protein by a filled circle. Apoptosis refers to the number of cells that commit to undergo cell death, and not to an augmentation of the basic cellular machinery by which this is accomplished. Checkpoints refer to highly patterned forms of cell cycle arrest in response to specific injurious agents. FA, genes of the Fanconi anemia core complex. TGFBRs and ACVRs, TGFß and activin receptors, types I and II. |
The TP53 gene (p53, chr. 17p) was identified as a protein bound by the major tumor antigen of the SV40 virus. Intragenic mutations were first found in colorectal cancer, and then in most types of malignancy (38,39). Next to p16, it is one of the most commonly mutated genes in human cancer. The mutations destroy the ability of the p53 protein to bind specific sequences of DNA (40), impairing its ability to stimulate the transcription of specific genes (41). These genes include the cell cycle inhibitor p21, which controls cell cycle checkpoints at the border of G1 and S (DNA synthesis) phases and in late G2 prior to M (mitosis) phase (42,43,44,45), and the 14–3-3σ protein, which maintains the late G2 checkpoint (46). These checkpoints involve the arrest of the cell division cycle in response to injurious exposures such as those resulting in DNA damage (47). p53 is also a powerful initiator of apoptotic mechanisms; the specific mechanism by which this is accomplished is not convincingly defined, but p53 is known to activate expression of the proapoptotic gene BAX (48). Fifty to 75% of pancreatic adenocarcinomas have intragenic mutations in TP53 (49,50).
The SMAD4 gene (DPC4/MADH4, chr. 18q), was identified in the search for the target gene of a hotspot of homozygous deletions affecting chromosomal arm 18q in pancreatic and biliary carcinomas (51). SMAD4 encodes a member of a family of Smad genes that are important in development and that transmit signals initiated by the binding of TGFβ-like proteins to cell surface receptors (52,53). Signal transduction may require the formation of complexes of Smad proteins, and both homooligomers and heterooligomers of Smad4 are described (54,55). Smad4 binds to specific sequences of DNA to stimulate the transcription of neighboring genes (56,57,58). It remains unclear which of these downstream genes serve as the effectors of SMAD4‘s tumor-suppressive effects; however, it is clear that the nuclear localization of Smad4 causes growth suppression and increased apoptosis (59). Missense mutations in the N-terminal half of the protein inactivate the ability to bind DNA, while those of the C-terminal half impair nuclear localization, activation of gene transcription, and probably the ability to form oligomers (54). Homozygous deletions and intragenic mutations of the SMAD4 gene occur in about half of pancreatic adenocarcinomas (51,60). Immunohistochemical analysis of Smad4 expression in tissues is an accurate means of identifying the tumors having genetic inactivation (Fig. 25.5) (61), in part because its missense mutations usually create an unstable protein (62,63).
The TGFβ and activin receptor genes encode a heterodimeric receptor pair comprising a type I receptor (for TGFβ, it is encoded by the TGFBR1/ALK5 gene; for activin, the ACVR1 and the ACVR1B/ALK4 genes) and a type II receptor (for TGFβ, the TGFBR2 gene; for activin, the ACVR2 and ACVR2B genes). TGFβ and activin are secreted proteins that serve as extracellular “growth factors”—ligands that bind to cell surface receptors. These receptors were first identified as TGFβ- and activin-binding proteins. Subsequently, mutations of the TGFBR2 gene, located at a polyadenine simple repeat within the gene, are nearly ubiquitous in cancers having microsatellite instability (MSI) and abnormalities of DNA-mismatch repair (64), and also, albeit at much lower frequencies, in non-MSI tumors of various organ sites (65). The ACVR2 gene accumulates similar mutations of a repeated sequence in MSI tumors (66,67). Genetic inactivation of the TGFBR1 gene was first shown in non-MSI pancreatic and biliary carcinomas (68). The binding of TGFβ and activin ligands to their receptors usually results in growth arrest or even apoptosis of cells; to accomplish this, both TGFβ and activin
receptors elicit the induction of expression of essentially similar sets of specific genes (69). These pathways can thus be considered as tumor suppressive. It is believed that to escape these effects, many cancers lose their responsiveness to TGFβ and activin. The type I receptors are kinases that phosphorylate Smad proteins 1–3. This event initiates the formation of Smad-Smad4 complexes, the nuclear localization of Smad4, and the induction of expression of downstream genes (70). The true relationship of TGFβ and SMAD4 tumor suppression is, however, still not precisely defined. In some cells, SMAD4 is necessary to convey the TGFβ-initiated growth arrest and transcriptional effects (71,72). In other cells, however, these actions of TGFβ persist even in the absence of SMAD4 (73,74). The coexistent genetic inactivation of both SMAD4 and a TGFβ receptor gene within individual tumors has also been described, suggesting that the products of these genes are not as closely linked in the same pathway as, for example, are p16 and Rb1 (68,75). Pancreatic adenocarcinomas with MSI usually have mutations of the polyadenine tract of both copies of the TGFBR2 gene and of the ACVR2 gene (19,67,68). Occasional non-MSI pancreatic and biliary carcinomas harbor homozygous deletions involving the TGFBR1 and TGFBR2 genes (68).
receptors elicit the induction of expression of essentially similar sets of specific genes (69). These pathways can thus be considered as tumor suppressive. It is believed that to escape these effects, many cancers lose their responsiveness to TGFβ and activin. The type I receptors are kinases that phosphorylate Smad proteins 1–3. This event initiates the formation of Smad-Smad4 complexes, the nuclear localization of Smad4, and the induction of expression of downstream genes (70). The true relationship of TGFβ and SMAD4 tumor suppression is, however, still not precisely defined. In some cells, SMAD4 is necessary to convey the TGFβ-initiated growth arrest and transcriptional effects (71,72). In other cells, however, these actions of TGFβ persist even in the absence of SMAD4 (73,74). The coexistent genetic inactivation of both SMAD4 and a TGFβ receptor gene within individual tumors has also been described, suggesting that the products of these genes are not as closely linked in the same pathway as, for example, are p16 and Rb1 (68,75). Pancreatic adenocarcinomas with MSI usually have mutations of the polyadenine tract of both copies of the TGFBR2 gene and of the ACVR2 gene (19,67,68). Occasional non-MSI pancreatic and biliary carcinomas harbor homozygous deletions involving the TGFBR1 and TGFBR2 genes (68).
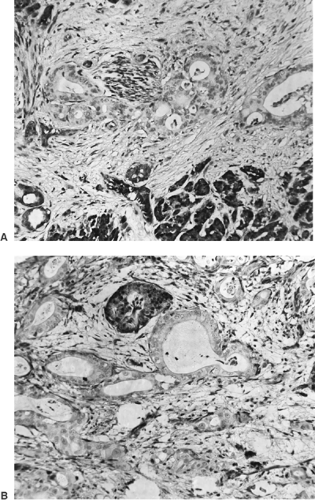 FIGURE 25.5. Immunohistochemistry accurately detects the genetic inactivation of the SMAD4 gene. This tumor had a homozygous deletion of the SMAD4 gene. A: Smad4 protein is immunolocalized within the epithelium of the remaining areas of pancreatic parenchyma (bottom), stromal fibroblasts (scattered), and within a nerve (upper center). Surrounding the nerve are the abortive duct structures of a moderately well-differentiated conventional type of pancreatic adenocarcinoma that lacks Smad4 immunoreactivity. A characteristic desmoplastic (scarring) reaction invests the area involved by carcinoma. B: A single pancreatic islet (upper center), expressing Smad4 protein, remains within a field of carcinoma lacking Smad4 expression. Monoclonal antibody B-8 (Santa Cruz) used with peroxidase detection, counterstained with hematoxylin. |
The MKK4 gene (MAP2K4, SEK1) was first identified as a member of the stress-activated protein kinase pathways that result in activation of Jun kinases and, although less well established, the p38 kinase (76,77). Activation of these pathways by exposure to ultraviolet light, hypoxia, certain cytokines and chemotherapeutic agents, etc., can result in apoptosis or differentiation. It was later found that a site of homozygous deletion, identified first in a pancreatic cancer, encompassed the MKK4 gene (78). Homozygous deletions or intragenic mutations of the MKK4 gene occur in about 4% of pancreatic cancers and in a similar fraction of other tumor types (78,79,80).
The STK11 gene (LKB1) was identified from a search for the mutated gene that causes Peutz-Jeghers syndrome (81,82). It encodes a serine-threonine kinase that activates AMP-activated protein kinase–related kinases and their downstream effects, including inhibition of the proproliferative mTOR pathway (83,84). Peutz-Jeghers syndrome has been shown to be associated with nearly a 24-fold elevated risk of pancreatic cancer (85). Homozygous deletions or intragenic mutations involving STK11 occur in about 4% of sporadic pancreatic and biliary carcinomas (86). In addition, loss of the wild-type allele was observed in a pancreatic cancer that arose in a patient with Peutz-Jeghers syndrome (86).
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


