CHAPTER 76 Other Inherited Metabolic Disorders of the Liver
Inborn errors of metabolism encompass a vast variety of disorders with myriad presentations including liver disease and are characterized by complex pathophysiology. Metabolic liver diseases may manifest as acute, life-threatening illnesses in the neonatal period or as chronic liver disease in adolescence or adulthood, with progression to liver failure, cirrhosis, or hepatocellular carcinoma. In the 2007 report of the Scientific Registry of Transplant Recipients in the United States, 3% of all liver transplantations in the United States were performed because of complications resulting from metabolic liver disease.1 When the pediatric population alone is analyzed, this percentage is substantially higher. Nationwide, more than 20% of the liver transplantations performed over the five-year period from 1995 to 2000 were for complications of metabolic liver disease.2 Liver transplantation has become a life-saving measure for many patients with metabolic liver diseases. New nontransplant treatment options have become available that may, in certain cases, obviate the need for liver transplantation and thereby help alleviate the shortage of donor organs.3,4
CLINICAL FEATURES OF METABOLIC LIVER DISEASE
The diverse presenting symptoms of metabolic liver disease are listed in Table 76-1. Certain metabolic liver diseases in young patients may mimic other illnesses, such as acute infections and intoxications. By contrast, the older patient with metabolic liver disease may present with symptoms of chronic disease. Because metabolic diseases can resemble multiple other disorders, a high index of suspicion is required for correct diagnosis.
Table 76-1 Presenting Features of Metabolic Liver Disease
| Symptoms | Coma |
| Developmental delay | |
| Growth failure | |
| Hyperammonemic symptoms | |
| Hypoglycemic symptoms | |
| Neurologic or motor skill deterioration | |
| Recurrent vomiting | |
| Seizures | |
| Signs | Ascites |
| Abdominal distention | |
| Cardiac dysfunction | |
| Cataracts | |
| Dysmorphic features | |
| Hepatomegaly | |
| Hypotonia | |
| Jaundice | |
| Short stature | |
| Splenomegaly | |
| Unusual odors | |
| Other Findings | Acidosis |
| Cholestasis | |
| Coagulopathy | |
| Fulminant hepatic failure | |
| Ketosis | |
| Rickets |
An infant presenting with cholestasis should undergo an evaluation for metabolic liver disease. (The approach to the patient with jaundice is discussed in detail in Chapter 20.) Any patient with progressive neuromuscular disease, developmental delay, or regression of developmental milestones also requires evaluation. Metabolic liver disease should be an immediate consideration in patients with elevated serum aminotransferase levels, hepatomegaly, acidosis, hypoglycemia, ascites, bleeding diathesis, hyperammonemia, coma, recurrent vomiting, or poor weight gain.
A detailed history can often raise the possibility of metabolic liver disease and help direct the investigation. A family history of consanguinity, multiple miscarriages, or early infant deaths may suggest a metabolic derangement. Close relatives with undiagnosed liver disease, progressive neurologic or muscle disease, or undiagnosed developmental delays should also raise suspicion. A carefully obtained dietary history is also important in dissecting the nature of the illness; introduction of certain foods may correlate with the onset of symptoms, as in patients with urea cycle defects, galactosemia, or fructosemia. A history of specific dietary aversions may also be revealing.5
Recommended initial screening tests, which should direct further diagnostic evaluation, are listed in Table 76-2. Because patients with metabolic liver disease often present with acute and recurrent symptoms, it is of utmost importance that the physician obtain the diagnostic studies as soon as possible and at the time the patient is experiencing symptoms. The laboratory values for many of these illnesses may normalize between acute episodes. In enigmatic cases, serum and urine samples should be obtained during the acute illness and saved (frozen) for definitive studies, if available. A liver biopsy can also be a valuable diagnostic tool. If a metabolic liver disease is suspected, in addition to obtaining specimens for standard histology, a frozen specimen should be saved and a sample prepared for later electron microscopic study to look at the subcellular organelles, which may exhibit characteristic changes in some disorders.
Table 76-2 Screening Laboratory Studies for Metabolic Liver Disease*
| Serum | Albumin |
| Alkaline phosphatase | |
| Amino acids | |
| Aminotransferases | |
| Ammonia | |
| Anion gap calculation | |
| Coagulation profile | |
| Electrolytes | |
| Ferritin | |
| Fractionated bilirubin | |
| Gamma glutamyl transpeptidase | |
| Glucose | |
| Lactate† | |
| Peripheral blood smear | |
| Pyruvate† | |
| Uric acid† | |
| Urine | Organic acids |
| Orotic acid | |
| Reducing substances |
* Specimens of serum and urine obtained during acute episodes should be saved for later studies.
α1-ANTITRYPSIN DEFICIENCY
Deficiency of α1-antitrypsin (α1-AT) is transmitted in an autosomal recessive fashion and leads to an increased risk of lung and liver disease. This deficiency is one of the most common genetic diseases in the world and the second most common metabolic disease affecting the liver, the most common being hereditary hemochromatosis (see Chapter 74).6,7 The following discussion focuses on the effects of α1-AT deficiency on the liver.
Pathophysiology
The prototypical member of the serpin family of protease inhibitors, α1-AT binds with and promotes the degradation of serine proteases in the serum and tissues. The most important of these serine proteases is neutrophil elastase, which is inhibited by α1-AT through formation of a tight 1 : 1 α1-AT-to-elastase complex. Loss of serum α1-AT activity, as occurs in the most common form of α1-AT deficiency, leads to uninhibited neutrophil elastase activity and is one of the primary mechanism for the premature development of pulmonary emphysema in affected patients.8 Another proposed mechanism derives from the observation that α1-ATZ polymers are proinflammatory toward neutrophils and are found in lung lavage fluid of patients with the protease inhibitor (Pi) ZZ phenotype of α1-AT deficiency (see later) and emphysema; moreover, instillation of α1-ATZ polymers in murine lung leads to an influx of neutrophils to the lung.8,9
Allelic α1-AT mutant variants produce Pi gene products that can be distinguished from the normal product by electrophoretic methods; the normal allelic representation is designated PiM. The PiZ variant produces a mutant α1-ATZ protein. Homozygosity at the PiZ allele is the most common and classic pathologic form of α1-AT deficiency and is capable of leading to liver and lung disease. The α1-ATZ molecule represents a single amino acid replacement of glutamine with a lysine residue as a result of a mutation at position 342 of the α1-AT (SERPINA1) gene. More than 100 naturally occurring variants of α1-AT have been described. Although most of these variants are either of no clinical significance or are extremely rare, two additional variants, PiSiiyama and PiMmalton, have been reported to be associated with liver injury and even cirrhosis.8,10–12
α1-AT is produced almost exclusively in the rough endoplasmic reticulum (ER) of hepatocytes and is subsequently targeted to the secretory pathway via the Golgi apparatus. Structural misfolding and polymerization of the mutant α1-ATZ protein causes its aberrant retention in the ER, failure of progression through the secretory pathway, and diminished intracellular degradation. In persons with the phenotype PiZZ, serum α1-AT activity levels are reduced to less than 15% of normal; this loss of function accounts for the development of pulmonary disease. Studies suggest that the rate of intracellular degradation may itself be genetically determined and may influence the expression of disease; α1-ATZ appears to be degraded more slowly in the ER of PiZZ patients who are susceptible to liver disease than in PiZZ patients who are not susceptible to liver disease.13
The exact mechanism for α1-ATZ-induced liver injury is not known. Studies of transgenic mice that express the human ATZ gene suggest a gain-of-function mechanism by which retention in the ER and accumulation in hepatocytes of mutant α1-ATZ is responsible for hepatotoxicity.14 Multiple intracellular signaling pathways, including caspase activation, ER stress responses, and the autophagic response, are activated by the retention of malformed proteins in the ER.15 Autophagy is an intracellular degradative pathway that targets proteins and organelles for destruction during development as well as at times of stress or nutrient deprivation. One hypothesis under investigation is that increased stimulation of autophagy by accumulation of α1-ATZ protein leads to ongoing mitochondrial injury, cellular apoptosis, and a chronic cycle of hepatocellular death and regeneration that may, over time, lead to liver injury and cirrhosis.7,16 Other genetic and environmental modifiers of the ER “quality control” process for handling mutant α1-ATZ protein are undoubtedly involved and account for the wide variation in clinical phenotype observed in the hepatic presentation of PiZZ patients.7
Clinical Features
Although the prevalence of the classic α1-AT deficiency allele, PiZ, is highest in populations derived from northern European ancestry, many racial subgroups are affected worldwide, and millions of persons have combinations of deficiency alleles (i.e., PiSS, PiSZ, or PiZZ).8,17 In the United States, the overall prevalence of deficiency allele combinations is approximately 1 in 490 (i.e., 1 in 1058 for PiSS, 1 in 1124 for PiSZ, and 1 in 4775 for PiZZ).18 Mounting evidence suggests that heterozygous α1-AT deficiency states can contribute to the development of cirrhosis and chronic liver failure in adults through mechanisms similar to those encountered with the PiZZ phenotype.10–1219 In addition, the heterozygous α1-AT deficiency state may contribute to worsening of chronic liver disease caused by hepatitis C viral infection or nonalcoholic fatty liver disease in adults, as well as cholestatic liver diseases in children.7,19–22
In the most unbiased study to date, reported by Sveger,23 on the epidemiology of liver disease in patients with α1-AT deficiency, 200,000 Swedish infants were screened for α1-AT deficiency, 184 infants were found to have abnormal allelic forms of α1-AT (127 PiZZ, 2 PiZnull, 54 PiSZ, and 1 PiSnull), and 6 (5 PiZZ and 1 PiSZ) died in early childhood, although only 2 from cirrhosis.23 Although about 10% of newborns with α1-AT deficiency (PiZZ) present with cholestasis and as many as 50% continue to have elevated serum aminotransferase levels at age three months, most are clinically asymptomatic.23,24 Liver disease does not develop in patients with null α1-AT phenotypes, whereas early-onset emphysema develops in all of them.25 Therefore, the prognosis for patients with liver disease manifesting in infancy as a result of α1-AT deficiency (PiZZ) is highly variable. Even those children in whom cirrhosis develops can have a highly variable progression to end-stage liver disease (ESLD), which infrequently leads to liver transplantation.26 Moreover, siblings with PiZZ have a variable degree of liver involvement; in a study reported by Hinds and colleagues, five of seven children with PiZZ requiring liver transplantation had siblings with PiZZ who had no persistent liver involvement.27 This finding suggests that environmental or additional genetic factors must be involved in determining the severity of liver disease in α1-AT deficiency; this area is under active scientific investigation.28
Of 150 patients with α1-AT deficiency from Sveger’s original study23 who subsequently underwent evaluation at age 16 and 18 years, none had clinical signs of liver disease. Elevated serum aminotransferase or gamma glutamyl transpeptidase (GGTP) levels were found in fewer than 20% of patients with a PiZZ phenotype and in fewer than 15% of those with a PiSZ phenotype.24 By the third decade of life, analysis of this same cohort of affected persons showed that 6% of PiZZ and 9% of PiSZ patients had a marginal increase in serum alanine aminotransferase levels.29
Even though liver disease is often (but not always) mild during infancy and childhood, patients with α1-AT deficiency have an increased risk for development of cirrhosis during adulthood; 42% of all PiZZ patients have histologic evidence of cirrhosis at autopsy.30,31 Moreover, homozygous α1-AT deficiency raises the risk for development of hepatocellular carcinoma, especially in men older than 50 years.31 The diagnosis of α1-AT deficiency should be considered in any patient presenting with noninfectious chronic hepatitis, hepatosplenomegaly, cirrhosis, portal hypertension, or hepatocellular carcinoma.
Diagnosis
α1-AT is considered a hepatic acute-phase reactant, and its release may be stimulated by stress, injury, pregnancy, or neoplasia. Because these factors can influence α1-AT production, even in patients with the PiZZ phenotype, the diagnosis of α1-AT deficiency should be based on phenotype analysis and not solely on the serum α1-AT level.32 A liver biopsy specimen, although not universally recommended, should confirm the diagnosis. Commercial tests are available to detect the most common mutant alleles by polymerase chain reaction analysis of genomic DNA. In addition, a re-sequencing molecular array chip is available for rapid sequencing of the entire SERPINA1 gene to allow identification of rare mutant alleles.33
Screening guidelines to diagnose α1-AT deficiency in asymptomatic persons have been proposed in an effort to reduce the risk of emphysema by counseling patients to avoid smoking.34 Adults with chronic lung disease and siblings of affected patients with lung or liver disease should be targeted for screening, and appropriate education and genetic counseling should be offered to patients with α1-AT deficiency identified by screening.
Treatment
The initial treatment of α1-AT deficiency is with symptomatic care. Breast-feeding until the end of the first year of life has been suggested to decrease the manifestations of cholestatic disease, as may the use of ursodeoxycholic acid.35 The importance of providing fat-soluble vitamins, when indicated, adequate nutrition, and counseling to avoid smoking and second-hand smoke cannot be overemphasized. The role of neonatal screening for α1-AT deficiency is still not settled, although the effect on smoking practices in patients diagnosed at an early age appears to be positive. If effective therapy for liver disease caused by α1-AT deficiency becomes available, neonatal screening may become more useful for preventing the need for liver transplantation.
Although progression to ESLD is uncommon, α1-AT deficiency is the most common metabolic liver disease for which liver transplantation is performed. Besides replacing the injured organ, transplantation corrects the metabolic defect, thereby preventing further progression of systemic disease. Between 1995 and 2004, 161 children and 406 adults underwent liver transplantation for ESLD associated with α1-AT deficiency in the United States; of these, 4.4% of the children and fewer than 1% of the adults were African Americans. Five-year patient survival rates following liver transplantation for the pediatric and adult patients with α1-AT deficiency were 83% and 90% respectively.36 Liver transplantation with grafts from donors with unrecognized α1-AT deficiency appears to have a comparable outcome to transplants using grafts without unrecognized liver disease.37
Replacement therapy with purified α1-AT is the only treatment option approved by the U.S. Food and Drug Administration (FDA) for lung disease associated with α1-AT deficiency. Patients who received replacement therapy in several studies, including a small randomized placebo-controlled trial, have had a slower rate of decline in lung tissue and function compared with control groups, although clinical efficacy has yet to be conclusively demonstrated.38 This therapy would not be expected to benefit α1-AT deficiency–associated liver disease, which results from malprocessed α1-ATZ, not loss of α1-AT protein function.
Other mechanistically based treatment options aimed at influencing the stability or secretion rates of α1-ATZ within the hepatocyte ER are being investigated. Chemical chaperones such as phenylbutyric acid (PBA) markedly increase secretion of α1-ATZ in experimental in vitro and in vivo models of α1-AT deficiency.39 A small pilot study investigated the potential benefits of PBA in the treatment of children with α1-AT–deficient liver disease. Unfortunately, no statistically significant increase in serum α1-AT levels occurred in PBA-treated patients, many of whom experienced unacceptable side effects.40 Another pharmacologic approach in the early stages of investigation involves the design of small molecules to inhibit α1-ATZ protein polymerization and increase clearance of intracellular aggregates.41
α1-AT deficiency is one of many diseases for which reconstitution of the normal genotype through gene therapy is being studied. Long-term expression of human α1-AT in murine liver, at therapeutic levels, has been achieved after hydrodynamics-based intravenous injection of nonviral deoxyribonucleic acid (DNA) constructs.42 Also, delivery of a vector carrying a ribozyme designed to target human α1-ATZ messenger ribonucleic acid (mRNA) in the portal vein of transgenic mice that carry the human PiZ allele led to a 50% reduction in α1-ATZ mRNA levels. In a separate experiment, administration of a vector carrying the normal PiM allele, which is resistant to ribozyme cleavage, led to long-term expression of human α1-AT protein in both liver and serum up to one year following injection.43 These results are promising first steps toward the goal of achieving successful gene replacement therapy for α1-AT deficiency.
GLYCOGEN STORAGE DISEASES
More than 10 distinct inborn disorders of glycogen metabolism have been described in the literature, but only 3 are associated with significant liver disease–glycogen storage disease (GSD) types I, III, and IV.44,45 Other GSDs may cause hepatomegaly or liver histologic changes but generally do not cause clinically important liver disease. The overall incidence of GSD types I, III, and IV is estimated to be between 1 in 50,000 and 1 in 100,000 population.
Glycogen metabolism occurs in many tissues, but the areas of clinical importance are the muscle and liver. The body uses glycogen as a storage system for glucose and as a ready reserve for times when systemic glucose is required (see Chapter 72). Glycogen is composed of long-chain glucose molecules arranged in a linear 1,4 linkage. From 8% to 10% of the glucose molecules are attached in a 1,6 linkage to form branching chains, which permit efficient storage of glucose while minimizing the impact on intracellular osmolality. The substrates for glycogen synthesis, glucose-6-phosphate (Glu-6-P) and glucose-1-phosphate (Glu-1-P), are derived from several pathways, including fructose and galactose metabolic cycles as well as gluconeogenesis and glycogenolysis (Fig. 76-1).
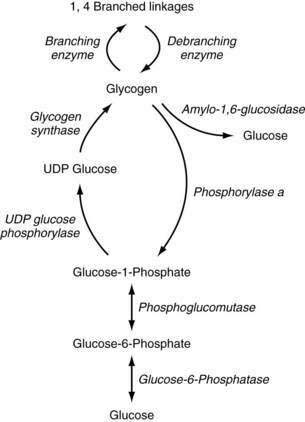
Figure 76-1. Pathway of glycogen synthesis and glycogenolysis. Enzymes are shown in italics. UDP, uridine diphosphate.
Through the action of uridine diphosphate glucose (UDPG) pyrophosphorylase and glycogen synthase, Glu-1-P is metabolized to UDPG and glycogen, sequentially. The 1,4 linkages can be converted to 1,6 linkages by the actions of branching enzymes. Amylo-1,6-glucosidase is a debranching enzyme that can release 8% to 10% of the glucose stored in glycogen. The remaining glucose is released as Glu-1-P through the action of phosphorylase a and is converted to Glu-6-P by phosphoglucomutase. Phosphorylase exists in an active (a) and an inactive form (b); protein kinase is responsible for the conversion of phosphorylase b to a. Protein kinase is stimulated by epinephrine, glucagon, and fasting, and thereby increases glycogenolysis. High levels of glucose influence the conversion of phosphorylase a back to phosphorylase b, thereby decreasing glycogenolysis. Glycogen synthase also exists in active (a) and inactive (b) forms. Phosphorylase a inhibits the conversion to glycogen synthase a, thereby reducing glycogen synthesis. High levels of glycogen favor the formation of glycogen synthase b.45
TYPE I
GSD type I, the most common inborn error of glycogen metabolism, results from deficiency of a two-component enzyme system involved in the transport of Glu-6-P from the cytosol into the ER by Glu-6-P translocase and subsequent cleavage of Glu-6-P by glucose-6-phosphatase (Glu-6-Pase), located on the luminal side of the ER. Clinical and molecular genetic observations have disclosed two subtypes of GSD type I, Ia and Ib, that account for virtually all cases.46 The clinical phenotype with respect to liver disease is similar in the two forms; however, patients with GSD type Ib often have intermittent severe neutropenia and neutrophil dysfunction, making them prone to recurrent episodes of severe bacterial infections and Crohn’s-like intestinal disease.47,48
Clinical Features
Most patients with GSD type I present in infancy with symptoms of metabolic derangement, such as lethargy, seizures, or coma as a result of profound hypoglycemia or metabolic acidosis, a protruding abdomen caused by hepatomegaly, muscular hypotonia, and delayed psychomotor development.49
Physical signs invariably include hepatomegaly, usually with a normal-sized spleen. Patients in whom the disease is poorly controlled for a long time exhibit short stature and growth failure and may be prone to adiposity. Delayed bone age and reduced postpubertal bone mineral density are common.50 Xanthomas can appear after puberty and localize to the elbows, knees, buttocks, or nasal septum, the last leading to epistaxis. Patients with GSD type I are susceptible to a wide spectrum of brain damage that may result in epilepsy, hearing loss, and abnormal neuroradiologic findings, most likely as a result of recurrent episodes of hypoglycemia.51
Other metabolic derangements can be seen. Lactic acid levels can reach four to eight times normal; the accompanying metabolic acidosis may manifest as muscle weakness, hyperventilation, malaise, headache, or recurrent fever. Serum adenosine triphosphate (ATP) and phosphorus levels are low, secondary to an increase in purine synthesis and the inability to release phosphorus from Glu-6-P. Hyperuricemia is common and may lead to gout, arthritis, or progressive nephropathy. Nephromegaly secondary to increased glycogen deposition is common, and with advancing age, progressive renal disease, hypertension, and renal failure requiring dialysis and transplantation may develop.49 Because of hypoglycemia, patients have chronically high serum levels of glucagon with depressed levels of insulin. Hypertriglyceridemia and hypercholesterolemia are present in both GSD Ia and GSD Ib (but more prominently in GSD Ia) and may account for the greater frequency of xanthoma formation.52
In addition to the features already noted, patients with GSD type Ib often have severe intermittent neutropenia and neutrophil dysfunction as well as high platelet counts. Crohn’s-like inflammatory bowel disease often occurs in patients with GSD type Ib at the time of severe neutropenia, and patients are prone to the development of severe bacterial infections, with abscess formation at numerous locations throughout the body.47 No correlation has been found between the severity of bacterial infections or degree of neutropenia and the molecular defect in Glu-6-P transport activity in humans with GSD type Ib, suggesting that other as yet unknown factors, such as modifying genes, define the ultimate disease phenotype.53
Hepatic Involvement
Hepatomegaly in GSD type I results from increased glycogen storage in the liver as well as a large degree of fatty infiltration; the latter likely develops because of a wide array of perturbations in lipid metabolism, including increased free fatty acid flux into the liver.52 Patients demonstrate mild elevations in serum aminotransferase levels but generally do not develop cirrhosis or liver failure.
Hepatic adenomas develop in 22% to 75% of patients, as early as three years of age but most commonly in the second decade of life, and tend to increase in both size and number as the patient ages (see Chapter 94).54 Adenomas in rare instances can transform to hepatocellular carcinoma; unfortunately, serum α-fetoprotein and carcinoembryonic antigen levels and features of the lesions on hepatic imaging are not predictive of malignant transformation.54–56 In the past, reports suggested that the risk of hepatic adenoma was increased in patients who were nonadherent with treatment of GSD type I, but more recent reports have found no differences in the risk of adenoma formation between adherent and nonadherent patients.57 In some patients, hepatic adenomas have been demonstrated to regress and disappear after adequate nutritional therapy, but in general, the course is unpredictable, especially in nonadherent patients.54,55 Because of the unreliability of radiographic imaging and serum marker levels in predicting malignant transformation in this patient population, the better approach—resection of an adenoma or liver transplantation—is uncertain.58
Diagnosis
The hepatic glycogen content is elevated in patients with GSD type I, and the most accurate diagnostic measure is direct analysis of enzyme activity performed on fresh, rather than frozen, liver tissue. Analysis of fresh liver tissue is important to avoid disrupting microsomal Glu-6-Pase activity.59 Fasting serum glucose and lactate levels, a positive glucagon response test result, and the response to fructose or galactose administration (patients with GSD type I do not show the expected rise in serum glucose concentration after administration of glucagon or either sugar) often provide supportive evidence but may not yield a definitive diagnosis. Intermittent severe neutropenia is noted in most patients with GSD type Ib.47,53 DNA analysis-based approaches that integrate biochemical features and the presence or absence of persistent neutropenia have been proposed and may provide a diagnostic alternative to liver biopsy.60
Treatment
Patients with undiagnosed or undertreated GSD type I are at increased risk of death, usually from hypoglycemic comas, seizures, metabolic acidosis, or, in those with GSD type Ib, sepsis from neutropenia.49 Rarely, hepatocellular carcinoma is a cause of death. Management centers on preventing the acute metabolic derangements and potential long-term complications and enabling the patient to attain normal psychological development and a good quality of life.59,61
Consensus guidelines for the management of GSD type I have been proposed.59,61 Biomedical targets for good metabolic control include a preprandial blood glucose level higher than 63 to 77 mg/dL (3.5 to 4.3 mmol/L), urine lactate-to-creatinine ratio higher than 0.06, high-normal serum uric acid level, venous blood base excess higher than −5 mmol/L, bicarbonate level higher than 20 mmol/L, serum triglyceride level lower than 6 mmol/L, and body mass index (BMI) between 0 and 2 standard deviations from normal. In addition, for GSD type Ib, demonstrating a normal fecal α1-AT level is desirable (see Chapter 28).59,62 Because optimal glycemic control is not always possible and the risk of severe hypoglycemia is high if delivery of glucose is interrupted inadvertently, serum lactate levels should be kept at the high end of normal because lactate is an alternative fuel for the brain.
Nutritional supplementation has become the mainstay of therapy for GSD type I. Frequent, high-carbohydrate, daytime feedings, such as uncooked cornstarch, or continuous nighttime drip feedings, or both, allow the steady release of glucose and lead to improved metabolic control and normalized growth and development.52,62 A biochemical target is to maintain the serum glucose level above 70 mg/dL (3.9 mmol/L). Uncooked cornstarch in a dose of 2 g/kg every six hours (6 to 8 mg of glucose/kg/minute) has been suggested; however, alternative regimens have been implemented successfully.
Prophylaxis with antibiotics (e.g., trimethoprim-sulfamethoxazole) is recommended for patients with GSD type 1b and severe neutropenia or recurrent bacterial infections.61 Granulocyte colony stimulating factor (GCSF) has been used with success in patients with GSD type 1b to improve hematologic parameters and neutrophil function and reduce the morbidity associated with severe bacterial infections.63 Splenomegaly may worsen with GCSF therapy, and bone marrow aspiration before and during GCSF therapy may be prudent, given rare occurrences of acute myelogenous leukemia (AML) in patients with GSD type 1b.61 Both GCSF and inflammatory bowel disease raise the risk of osteopenia, and monitoring of bone density is advised.
Adenoviral-mediated gene replacement therapy of recombinant Glu-6-Pase in a canine model of GSD type Ia deficiency, which has all of the major features of GSD type 1a in humans, has led to encouraging results and may be an option in humans in the future.64 An alternative approach, human hepatocyte transplantation, has been performed in a single patient with GSD type 1a with near-resolution of hypoglycemic episodes; however, hypoglycemia subsequently recurred, likely because of the lack of ongoing immunosuppression to reduce the likelihood of recipient rejection of transplanted donor hepatocytes.65 Hepatocyte transplantation, with the use of standard post-transplantation immunosuppression, has been performed successfully in an 18-year-old male adolescent with GSD type 1b, with euglycemia maintained up to 2 years post-transplantation.66
Liver transplantation has corrected the metabolic error in patients with GSD type I and permitted catch-up growth, even in patients in the third decade of life.67,68 Neutrophil counts and function, however, are only variably improved after liver transplantation in patients with GSD type 1b.67
TYPE III
GDE is encoded by a single gene and possesses two independent catalytic activities, an amylo-1,6-glucosidase and oligo-1,4→1,4 glucan transferase. Both of these activities are deficient in the two main clinical subtypes of GSD type III, types IIIa and IIIb. Differential expression of four major GDE mRNA isoforms in liver and muscle tissue distinguishes the two types: type IIIa affects liver and muscle and accounts for 80% of patients, and type IIIb affects the liver only and accounts for 15% of patients. Although subtype-specific mutations in the GDE gene are increasingly identified, the molecular basis for this differential tissue-specific expression of GDE is unknown, and no clear genotype-phenotype correlations have been identified.44,69 Rare isolated loss of one of the two GDE activities has been observed (i.e., glucosidase activity in type IIIc and transferase activity in type IIId).
Clinical Features
Patients with GSD type III may also display progressive muscle weakness, which worsens with activity, and muscle wasting. Nephromegaly is not seen, but cardiomegaly may be present. The diagnosis can be made by direct enzyme analysis of muscle or liver tissue or peripheral leukocytes; mutation analysis of the GDE gene will be increasingly important for diagnosis in the future.45,70 Hepatic adenomas develop in approximately 25% of patients, and isolated reports of cirrhosis leading to hepatocellular carcinoma have been reported.55,71
TYPE IV
Deficiency of the branching enzyme is seen in GSD type IV, a rare syndrome also known as amylopectinosis. Glycogen and amylopectin accumulate in hepatocytes, leading to hepatomegaly, abdominal distention, and failure to thrive, most commonly during infancy. Signs of liver disease, when present, predominate later in the course of the disease. Several variable forms of GSD type IV have been observed—a severe congenital form that manifests as fetal hydrops, neonatal hypotonia, or fetal death72; a childhood subtype that manifests as cardiomyopathy and abnormal neuromuscular development; and other milder, nonprogressive presentations of hepatic disease that do not lead to cirrhosis and are not associated with skeletal muscle or neurologic involvement.73 Genotype-phenotype analyses of the branching enzyme gene have revealed a high degree of molecular heterogeneity without clear clinical associations.73
Hypoglycemia is relatively uncommon, and responses to fructose and galactose challenges are normal. Serum lactate and pyruvate levels are normal, and aminotransferase levels are only moderately elevated until more severe liver involvement becomes apparent. Progressive macronodular cirrhosis is present with an abundance of PAS-positive deposits (amylopectin) in hepatocytes (Fig. 76-2). Cirrhosis may progress to liver failure, and adenomas and hepatocellular carcinoma may develop rarely.74
The diagnosis of GSD type IV can be made by direct enzyme analysis of liver tissue or fibroblasts. Most patients die within the first three years of life if the disease is untreated. Diets high in protein and low in carbohydrate have been associated with improved growth but have had little effect on liver involvement. Liver transplantation has been used successfully and results in correction of the metabolic error and normal growth; however, persistence of amylopectin deposits in the heart (with progressive cardiomyopathy) and leukocytes of affected patients has been described.75
CONGENITAL DISORDERS OF GLYCOSYLATION
Congenital disorders of glycosylation (CDGs) comprise a group of inherited defects in the enzymes that synthesize the glycan moiety of glycoproteins or the macromolecules that affect intracellular trafficking and functioning of glycoproteins.76–78 More than 20 CDGs involving both asparagine (N)- and serine/threonine (O)-linked protein glycosylation have been reported to date, and many of these disorders lead to dysfunction of the liver, intestine, or both.76–78 Protein glycosylation is complex and involves multiple enzymatic steps and subcellular compartments.77,78 Secretory glycoproteins with altered carbohydrate moieties in CDGs include coagulation factors, albumin and other binding proteins, growth hormone, apolipoproteins, insulin, and thyroxine-binding globulin.79 Because protein glycosylation occurs in all cells, it is not surprising that patients with a CDG exhibit multisystemic abnormalities, often dominated by central nervous system manifestations.
Two main groups of protein N-glycosylation disorders, groups I and II, have been delineated on the basis of characteristic isoelectric focusing patterns of serum transferrin, a marker protein for this group of disorders.77 Group I disorders (of which there are 12 types) involve aberrant processing of lipid-linked oligosaccharides before transfer to protein targeted for glycosylation and include the three most common and best-characterized types of CDG, types Ia, Ib, and Ic. Clinical features in common among these three disorders are protein-losing enteropathy, coagulopathy (both procoagulant and anticoagulant states), feeding difficulties, and hepatomegaly.77,80
CDG type Ia is caused by defects in phosphomannomutase (PMM), an enzyme that converts mannose-6-phosphate to mannose-1-phosphate (Fig. 76-3). Almost 60 mutations have been found in the encoding PMM2 gene; most patients are compound heterozygotes for mutations that likely preserve some residual PMM enzymatic activity, suggesting that complete loss of PMM activity is incompatible with life; indeed, targeted disruption of the PMM2 gene in the mouse leads to early embryonic lethality.81,82 Patients typically have severe neurologic abnormalities, dysmorphisms (inverted nipples, abnormal fat distribution, and esotropia), and congenital hepatic fibrosis and steatosis in addition to the features described earlier.80,83
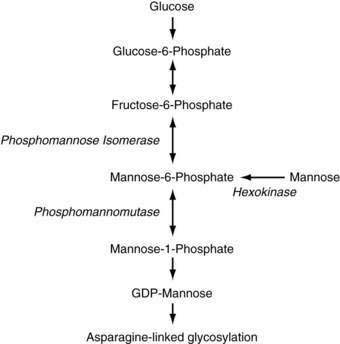
Figure 76-3. Pathway of mannose metabolism. Enzymes are shown in italics. GDP, guanosine diphosphate.
Patients with CDG type Ib have a defect in phosphomannose isomerase (PMI), which converts fructose-6-phosphate to mannose-6-phosphate. In addition to intractable diarrhea, protein-losing enteropathy, and congenital hepatic fibrosis, recurrent episodes of hyperinsulinemic hypoglycemia and cyclic vomiting can occur in patients with CDG type Ib. Neurologic symptoms are usually absent and dysmorphisms are less common than in CDG type 1a.76,80,84 Many patients with CDG type Ib have been treated effectively with dietary mannose, making CDG type Ib the only treatable form of CDG to date, although liver fibrosis can still develop despite improvement in clinical symptoms.78,85,86
Transient hepatomegaly, without congenital hepatic fibrosis, has been noted in a patient with CDG type Ic; otherwise, the clinical features of CDG type Ic are similar to but milder than those of CDG type Ia.80 Patients with CDG type Ih also have protein-losing enteropathy with chronic diarrhea, hepatic fibrosis, and variable degrees of hepatic dysfunction.87 Children with untyped cases of CDG, labeled CDG-X until the genes and protein products can be identified, have been found and have isolated cryptogenic chronic liver disease, mild coagulopathy, and mild portal fibrosis and focal steatosis on liver biopsy specimens.88 In some cases, numerous myelinosomes have been identified in hepatocytes by electron microscopy.
Group II CDGs (six types) involve defects that affect the processing of N-linked glycoproteins.77,78 Most types result from defects in enzymes involved in the trimming of protein-bound oligosaccharides and the subsequent addition of terminal sugars. Patients have marked dysmorphic features and severe developmental retardation. Two infants with hepatosplenomegaly, progressive jaundice, severe epilepsy, recurrent infections, and heart failure have been shown to have mutations in a subunit of the conserved oligomeric Golgi (COG) complex that result in disruption of glycosylated protein intracellular trafficking.89 Another CDG type II disorder with liver involvement has been reported in an infant with recurrent infections, chronic diarrhea, progressive cirrhosis, and normal hepatic excretory and synthetic function.89 The biochemical and clinical features of the remaining types of CDG are less well characterized.
Isoelectrofocusing of apolipoprotein C-III, which carries a single O-linked glycan moiety, has been proposed as a screening method for the O-glycan biosynthesis defects (six types) encountered thus far.76,90 Hepatic dysfunction is usually mild in CDG and usually does not lead to symptoms. Mild hepatic steatosis and fibrosis typically are seen on light microscopy; on electron microscopy, lysosomal vacuoles, termed myelosomes, with concentric electron-dense membranes and variable electron-lucent and electron-dense material, are noted.91 Patients uncommonly can progress to liver failure, with micronodular cirrhosis noted at autopsy.
Any patient with unexplained congenital hepatic fibrosis, protein-losing enteropathy, or a procoagulant tendency should be evaluated for the possibility of CDG (especially type Ib). An initial screening of serum transferrin with isoelectric focusing should be performed, followed by confirmatory enzymatic analysis in fibroblasts, leukocytes, or liver tissue. If the diagnosis of CDG type Ib is confirmed, oral mannose therapy should be initiated.86
PORPHYRIAS
Pathophysiology
The metabolic pathways of heme synthesis are essentially the same in the two tissues in which heme synthesis primarily occurs, the liver (15% to 20%) and the bone marrow (75% to 80%), although synthetic control may be different in the two tissues. The rate-limiting step in hepatic heme synthesis begins with the conversion of glycine and succinyl coenzyme A (CoA) to 5-aminolevulinic acid (ALA) by the action of ALA synthase (Fig. 76-4). ALA synthase activity is decreased by the end-product of the pathway, heme, and is increased by substances that induce the hepatic cytochrome P450 pathway. Six additional enzymatic steps convert ALA to protoporphyrin IX (see Fig. 76-4). In the final step of the pathway, protoporphyrin IX is coupled to ferrous iron by ferrochelatase to create heme. Enzyme deficiencies arising from any of these eight steps of the heme synthetic pathway lead to the clinically apparent diseases known as the porphyrias.
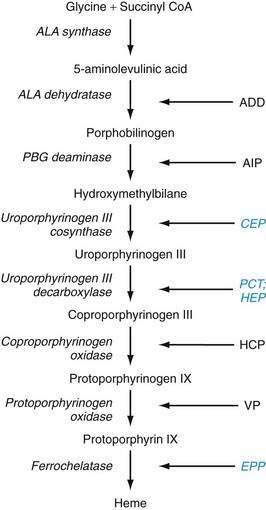
Porphyrias are commonly classified according to clinical features into two main groups, acute porphyrias, which are characterized by dramatic and potentially life-threatening neurologic symptoms, and cutaneous porphyrias, which typically cause few or no neurologic symptoms but instead give rise to a variety of severe skin lesions (Table 76-3). In five of the porphyrias, the liver is the major site of expression; in two others, both the liver and bone marrow are involved; and in one, only the bone marrow contributes.
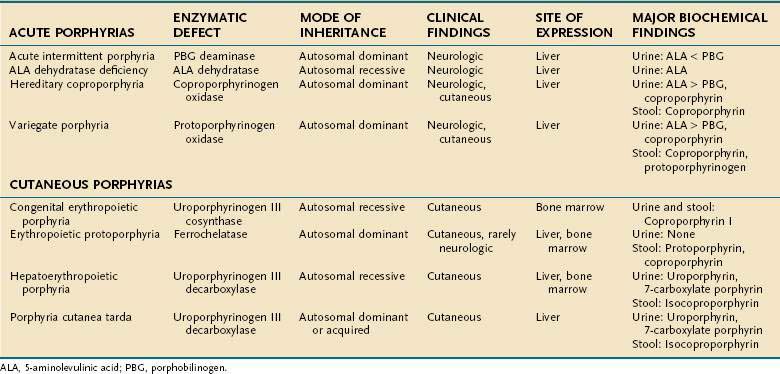
Acute Porphyrias
The symptoms and signs of the acute neurovisceral attacks that occur in the four acute porphyrias vary considerably. Abdominal pain is present in more than 90% of patients, followed in frequency by tachycardia and dark urine in about 80% of patients. Neuropsychiatric features include hysteria, depression, psychosis, confusion, hallucinations, seizures, and coma, although little evidence suggests that chronic psychiatric illness occurs. Other features are constipation, extremity pain, paresthesias, nausea, vomiting, urinary retention, hypertension, peripheral sensory deficits (often in a “bathing trunk” distribution), and weakness leading to ascending paralysis or quadriplegia. These neurologic attacks appear to be related to the overproduction of ALA and porphobilinogen (PBG), which leads to higher serum and tissue levels of these neurotoxic products.92
Acute episodes are about five times as common in women as in men and may be precipitated by many factors, most commonly drugs, alcohol ingestion, and smoking. Steroids, sex hormones, and medications that stimulate the hepatic cytochrome P450 system, perhaps by increasing the requirements for heme production, are often identified as precipitants.93 Other inciting factors are fasting, infections, and pregnancy; some women report greater problems during the luteal phase of their menstrual cycles.92,94 The disease is clinically latent in 65% to 80% of patients.
ALA dehydratase deficiency is a rare syndrome with autosomal recessive transmission in which the enzyme activity is less than 3%. The enzyme activity is 50% of normal in carriers, who are asymptomatic. Affected patients have severe, recurrent neurologic attacks that may be life threatening. They excrete large amounts of ALA in their urine. Liver transplantation was reported to result in complete resolution of symptoms in one patient with ALA dehydratase deficiency.95
The three remaining acute porphyrias—acute intermittent porphyria (AIP), hereditary coproporphyria (HCP), and variegate porphyria (VP)—result from partial deficiency of the enzymes PBG deaminase, coproporphyrinogen oxidase, and protoporphyrinogen oxidase, respectively. All three disorders are inherited in an autosomal dominant fashion with variable expression. AIP is the most common of the three conditions, occurring in 5 to 10 per 100,000 people, and manifests primarily as derangements in the autonomic nervous system or as a psychiatric disorder.96 VP is more common in South Africa than elsewhere. Although HCP and VP give rise to neurologic symptoms similar to those of AIP, cutaneous lesions also occur in HCP and predominate in VP.97
Cutaneous Porphyrias
The cutaneous porphyrias differ from the acute porphyrias in that affected patients exhibit few or no neurologic symptoms. In these illnesses, excess porphyrins or porphyrinogens are deposited in the upper dermal capillary walls, where these photoreactive compounds cause tissue damage that manifests as cutaneous vesicles and bullae in areas exposed to light or excessive mechanical manipulation. Scarring, infection, pigment changes, and hypertrichosis can follow and even lead to severe mutilation.98
Porphyria cutanea tarda (PCT), the most common of the porphyrias, typically involves a 50% reduction in activity of the enzyme uroporphyrinogen decarboxylase. Patients usually present after 20 years of age. Two types of PCT are recognized. Type I PCT affects 80% of patients and is a sporadic (acquired) form with the enzyme deficiency restricted to the liver. Type II, which affects the other 20% of patients, is familial and inherited in an autosomal dominant fashion with incomplete penetrance; the enzyme deficiency occurs in all tissues.99 Symptoms develop in fewer than 10% of patients with type II PCT. Type I PCT is associated strongly with high alcohol intake, estrogen therapy, and systemic illnesses, including systemic lupus erythematosus, diabetes mellitus, chronic kidney disease, and the acquired immunodeficiency syndrome. For unclear reasons, concomitant hepatitis C infection is strongly associated with expression of PCT (see Chapter 79).100,101 The frequency of mutations of the HFE gene, which causes hereditary hemochromatosis, is increased in patients with types I and II PCT, and these mutations are thus susceptibility factors for clinical expression of the PCT phenotype (see Chapter 74).100,102,103 This association is consistent with pathologic findings in liver biopsy specimens from patients with PCT, of whom 80% have siderosis, 15% have cirrhosis, and most have evidence of iron overload. A patient in whom hepatocellular carcinoma developed has been reported to have unsuspected underlying PCT and hereditary hemochromatosis with bridging fibrosis.104,105 Patients usually do not show signs of overt clinical liver disease, apart from elevated serum aminotransferase levels.
Hepatoerythropoietic porphyria (HEP) is a rare form of porphyria with a pathogenesis similar to that of PCT. HEP results from homozygous uroporphyrinogen decarboxylase deficiency, yielding less than 10% of normal enzyme activity. The cutaneous lesions, which resemble those of PCT, are typically severe and mutilating. The disease usually manifests in the first year of life, and as the patient ages, the dermatologic manifestations may subside, but liver disease, characterized by a nonspecific hepatitis, worsens. Mutation analysis of the uroporphyrinogen decarboxylase gene has revealed numerous mutations that are usually unique to an individual family; no clear genotype-phenotype correlations have been observed.99
Erythropoietic protoporphyria (EPP) is caused by partial deficiency of the enzyme ferrochelatase (FECH), the final step in the heme synthetic pathway. EPP is the second most common type of porphyria and is inherited in an autosomal dominant manner with variable penetrance. Patients with EPP and asymptomatic carriers exhibit an FECH enzyme activity of 30% to 40% and 50%, respectively, even though both groups inherit a defective FECH allele. The mechanism for the variable clinical expression is explained by co-inheritance of a “low-expressing” normal FECH allele in symptomatic patients with EPP and a normal FECH allele in asymptomatic patients.106,107
Clinical liver disease, which develops in 5% to 10% of patients with EPP, results from progressive hepatic accumulation of protoporphyrin.108 Liver disease typically occurs after age 30 but has been described in children. The liver appears black and nodular, with hepatocellular necrosis, portal inflammation, cholestasis, and extensive deposits of dark brown pigment in hepatocytes, Kupffer cells, and biliary structures; birefringence of pigment deposits is seen on polarization microscopy.109 Of 57 patients with EPP followed for more than 20 years in one study,110






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


