Approach to treatment
Given the chronic progressive nature of obesity, obesity management has to be approached with the same persever-ance and diligence as the management of other chronic diseases [7]. All behavioral, medical or surgical interven tions have to be continued beyond the weight-loss phase to prevent weight regain. Therefore, in a given patient, any intervention aimed at weight loss has to be reasonably sustainable in the long term to avoid recidivism or weight regain.
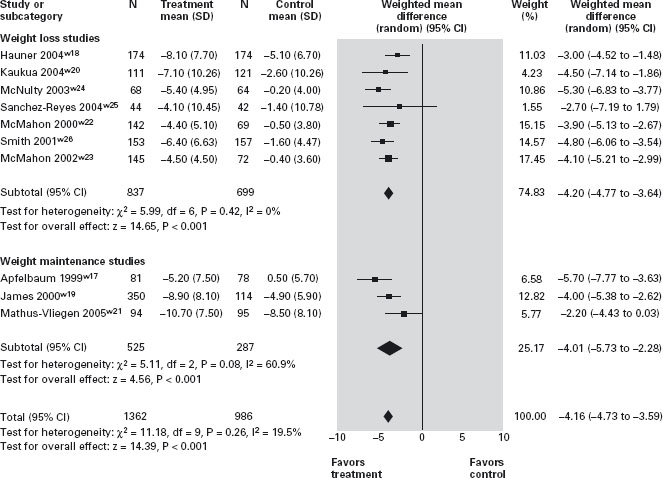
A wide range of behavioral interventions has been shown to be successful in the long term. These include self-monitoring with food journals and pedometers, regular weighing and regular provider contact [7]. Patients with emotional and binge-eating disorder may benefit from cog nitive behavioral therapy [16]. Failure to identify and manage binge-eating appropriately is associated with vir-tually 100% recidivism. Overall the long-term, weight-loss benefit of behavioral intervention is rather modest. In a recent multicentre study that randomized 1029 partici pants, who had lost an average of 8.5 kg through lifestyle change over six months, to various strategies for weight-loss maintenance (personal contact, unlimited access to an interactive technology-based intervention, or self-directed control) over 30 months, resulted in weight loss ranging between 2.9 to 4.2 kg at the end of the study [12]. A1
Pharmacological treatment for obesity should be consid ered in all patients who need to achieve a 5–10% weight loss [7]. In Canada and the USA there are currently two compounds licensed for the long-term treatment of obesity: orlistat, a gastrointestinal lipase inhibitor that reduces digestion and absorption of fat by around 30% [17] and sibutramine, a serotonin and norepinephrine reuptake inhibitor that increases satiety and may have modest effects on thermogenesis. Major adverse effects of orlistat include steatorrhea and oily rectal discharge. Use of sibutramine can be associated with xerostomia, consti pation, hyperhydrosis, insomnia, palpitation and, in rare cases, an increase in blood pressure [17]. Both compounds have been shown to significantly increase weight loss when added to moderately caloric-restrictive diets. More importantly, continuation of pharmacotherapy is associ ated with a more significant reduction in weight regain than behavioral treatment alone. Therefore, long-term use of these agents in patients who respond to and tolerate these drugs is recommended. Despite documented effects on obesity-related morbidity, it should be noted that thus far, there is no evidence that behavioral or pharmacologi cal therapy of obesity reduces overall or obesity-related mortality. A1a
Bariatric surgery is currently the most effective long-term treatment of severe obesity and has been shown to dramatically reduce the risk for co-morbidities including cardiovascular disease, type II diabetes, obstructive sleep apnea and certain cancers, and the risk for overall mortality alone (by up to 30%). Thus, for example, the recent Swedish Obesity Study was a prospective case control study com paring 2010 surgical to 2037 non-surgical matched controls [14]. During an average of 10.9 years of follow-up (follow-up rate, 99.9%) there were 129 deaths in the control group and 101 deaths in the surgery group (hazard ratio adjusted for sex, age, and risk factors was 0.71; p = 0.01). The most common causes of death were myocardial infarction (control group, 25 subjects; surgery group, 13 subjects) and cancer (control group, 47; surgery group, 29). These benefits were seen with average weight loss after 10 years of 25% (gastric bypass), 16% (vertical-banded gastro-plasty) and 14% (gastric banding). Currently, the most common surgical procedures are laparoscopic adjustable gastric banding (LABG) and laparoscopic Roux-en-Y gastric bypass (RYGB) surgery. While overall complication rates at experienced centers are low (1–2%), surgical com plications are even lower with LABG. However, weight loss with this procedure is less pronounced and is more dependent on patient compliance than is the case for RYGB. Other surgical approaches including sleeve gastrec-tomies and endoluminal approaches are currently under investigation.
Given the complexity of obesity management, interven tions are best provided by an interdisciplinary team of nurses, psychologists, physiotherapists, dieticians, social workers, occupational therapists, physicians and surgeons. In light of the widespread bias and discrimination of health professionals against obese clients, bariatric personnel should undergo sensitivity training [5].
Currently there is no cure for obesity – only long-term treatments that require ongoing support and follow-up of all patients to ensure adherence and reduce recidivism. Therefore, it is important to make every effort possible to prevent the development of this chronic and debilitating condition.
The role of the gastrointestinal tract in ingestive behavior
Appetite is regulated by a complex interaction between central and peripheral signals that affect both the sensation of hunger and how an individual responds to food. While older models suggested that hypoglycemia stimulates food intake and post-prandial hyperglycemia activates the satiety centre of the brain [18], in the last decade a much more complex system has emerged that involves a multi tude of neuropeptide and neurotransmitter systems, the enteroendocrine system and peripheral signals derived from adipose tissue (e.g. leptin).
The response to food ingestion is traditionally consid ered to occur in three phases. The cephalic phase is char acterized by the secretion of saliva, gastric acid and pancreatic enzymes that is triggered by visual and olfac tory stimulation. Relaxation of the sphincter of Oddi and the gastric fundus, and increased contractions of the gall bladder occur [19]. This cephalic phase is followed by the gastric phase, which involves relaxation of the proximal stomach. This is caused by the activation of inhibitory neurons on the gastric wall (accommodation). Finally, the intestinal or post-ingestion phase is characterized by a combination of duodenal, intestinal and colonic responses that may also play a role in satiety.
A multitude of enteroendocrine hormones or peptides play a role in regulating food intake by activating nearby extrinsic sensory fibers (vagal and spinal) or via the blood stream (Table 25.1) [20]. They generally have a short half-life, interact with other hormones and they can modulate the sensitivity of the vagal response. Through activation of the vagus nerve or through direct neural stimulation, these peptides stimulate the hypothalamus, which then inte grates these signals. The main site of integration is the arcuate nucleus of the hypothalamus (ARC). Two major sub-populations of neurons in the ARC are implicated in the regulation of feeding; cocaine and amphetamine related transcript (CART) and pro-opiomelanocortin (POMC) inhibit food intake, whereas neuropeptide Y (NPY) and agouti-related protein (AgRP) stimulate it. Both influence how the hypothalamus signals pituitary hormone secretion and appetite [21]. The neurophysiological pathways involved in triggering hunger and satiety are complex and beyond the scope of this chapter.
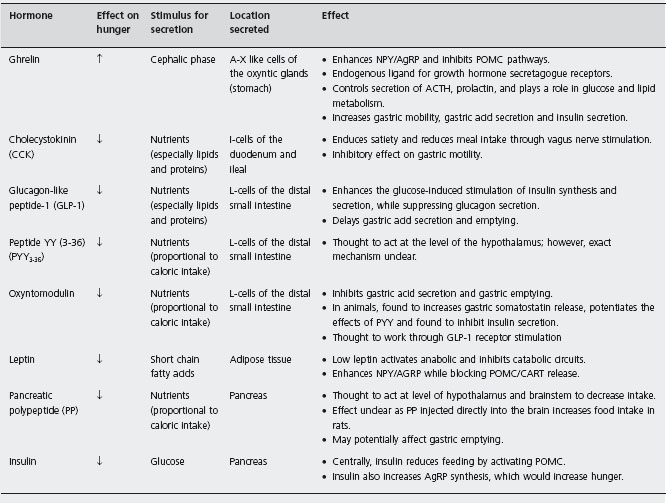
In the stomach, neural sensors perceive changes in the tension, stretch and volume of the stomach wall and outputs from these mechanoreceptors are relayed to the brain via the vagus and spinal sensory nerves [20] to the nucleus tractus solitarius (caudal brainstem) and then to the hypothalamus. Gastric distension induces a feeling of satiety [22]. Persistent delayed gastric emptying is known to decrease food intake, and artificial distension of the proximal stomach with a water-filled balloon has been found to reduce oral intake [23, 24]. On the other hand, the stomach is also the source of ghrelin, the only enteric hormone so far known to promote hunger and food intake.
In contrast, there are a number of enteric satiety signals, the best studied of which include cholecystokinin (CCK), glucagon-like peptide-1 (GLP-1) and peptide YY (PYY) [21]. Parental administration of CCK induces satiety in animals and humans [25,26,27], an effect that is eradicated by vagotomy. CCK also delays gastric emptying [28] result ing in gastric distension. Due to its short duration of action, attempts to utilize CCK as a treatment for obesity have failed. PYY, another satiety hormone released from the colon, also inhibits fasting small bowel motility and gastric emptying [29]. Parenteral PPY injection decreases food intake in normal and obese humans [30], potentially via a central mechanism. Exogenous GLP-1 has been found to decrease food intake [31], only when administered at doses much higher than those physiologically achieved after a normal meal [32].
Gastrointestinal symptoms and disease in the obese patient
Gastro-esophageal reflux disease (GERD)
Obesity is a significant risk factor for GERD. Obese indi viduals are more than twice as likely as non-obese indi viduals to admit to a history of GERD or symptomatic heartburn [33] [34]. The prevalence of GERD ranges from 8-26% in individuals with obesity [35] [36]. Similarly, endoscopically diagnosed erosive esophagitis is also more common in individuals with an increased BMI, with an odds ratio (OR) of 1.76 [37]. Several mechanisms have been described to explain this correlation. An increased intra-abdominal and resulting increase in intragastric pressure can precipitate reflux into the esophagus or hiatus hernia [38]. In addition, derangements of esophageal motility, including hypertensive contractions (nutcracker esopha gus) or disordered contractions (non-specific motility dis order), decreased lower esophageal sphincter pressure, inappropriate lower esophageal relaxation, a sliding hiatus hernia and delayed gastric emptying have also been impli cated (see motility section below) [39]. Finally, diet, reflux-ate composition and an increased incidence of Helicobacter pylori may also play significant roles in increasing the inci dence of GERD in obese individuals.
Several (but not all) studies suggest that weight loss is associated with decreased esophageal acid exposure and an improvement in GERD symptoms [40]. The lack of symptomatic improvement in many patients following weight loss may be attributed to the persistence of a hiatus hernia [41]. Modifications in dietary composition may be more effective than an actual change in weight [42, 43]. Symptomatic improvement following bariatric surgery is likely secondary to both weight loss and a change in the anatomy of the gastrointestinal (GI) tract. Roux-en-Y con sistently results in a favorable response as an anti-reflux procedure, with a significant improvement in reflux symp toms and a decrease in the use of GERD medication noted postoperatively. Adjustable gastric banding may not be a suitable option in obese individuals with symptomatic GERD, unless combined with anti-reflux surgery, since banding can precipitate the reflux of gastric contents into the esophagus from the small gastric reservoir [44].
Acute pancreatitis
Severe hypertriglyceridemia, not uncommon in overweight and obese individuals, is a rare but well recognized cause of acute pancreatitis [45]. Increased BMI is, however, con sidered to be a risk factor for a worse outcome in patients with acute pancreatitis [46, 47]. In a recent meta-analysis severe acute pancreatitis was more frequent in obese patients (BMI ≥ 30kg/m2) (OR 2.6), who also tended to develop more systemic (OR 2.0) and local complications (OR 4.3). Local complications from the greater accumula tion of peripancreatic fat included fat necrosis, with necro sis becoming a nidus for infection [46]. This is particularly problematic in obese individuals with hyperinsulinemia and insulin resistance [48]. Obesity is also associated with respiratory, renal and circulatory compromise as well as an increased incidence of death [49]. In light of these observa tions, adding an obesity score (BMI ≥ 26kg/m2) to the APACHE II (Acute Physiology and Chronic Health Evaluation) score has been recommended to improve its predictive value for overweight and obese patients [50, 51]. Thus, the composite APACHE-O (APACHE-II plus obesity) score >8 has greater predictive accuracy than the APACHE-II score, with a sensitivity of 82%, specificity of 86%, posi tive predictive value (PPV) of 74% and a negative predictive value of 91% (NPV) in predicting morbidity and mortality [50].
Gallstone disease and cholecystitis
Both obesity and rapid weight loss are associated with an increased risk of gallstone disease, particularly in women. Risk appears highest when obesity begins in late adoles cence [52] or if obesity is truncal [53]. The three major factors important in the development of gallstones include gall bladder dysmotilty, cholesterol hypersaturation of bile (i.e. a higher concentration of cholesterol to bile or phos-pholipids), and cholesterol nucleation. Obese patients have an increased biliary secretion of cholesterol from the liver, which leads to the formation of bile supersaturated with cholesterol and the precipitation of cholesterol microcrys-tals [54]. Both diabetes mellitus and insulin resistance are known to be confounding factors with respect to the asso ciation of obesity and gallstone disease [55]. Increased hepatic cholesterol synthesis and diets high in calories and refined carbohydrates, but low in fiber, also increase the risk. There is contradictory evidence, however, on the spe cific role of total fat intake [56]. There has been no correla tion found between serum cholesterol level and a predisposition to gallstone formation [57].
Impaired gall bladder emptying or gall bladder stasis, and further cholesterol saturation, as would occur in indi viduals who lose weight rapidly with restricted dieting or following bariatric surgery, also precipitates stone forma tion [58]. Low calorie and low fat diets affect biliary lipid composition with increases in the hepatic uptake of choles terol and hepatic cholesterol synthesis [59]. Weight loss is clearly associated with the development of gallstones, especially in women [51]. In several studies, approximately 11–28% of obese patients who severely restricted their dietary intake [60–62], and 27–43% of patients who under went bariatric surgery developed gallstones within a period of 1–5 months [63, 64]. Routine concomitant chole-cystectomy with bariatric surgery has been suggested, but remains controversial. The rate of weight loss also influ ences gallstone development. As gallstone formation appears particularly high when weight loss is more than 1.5kg per week [65], preventive strategies have been sug gested. These include controlling the rate of weight loss, administration of ursodeoxycholic acid to decrease choles terol saturation, limiting between meal fasting time and maintaining a small amount of fat in the diet [66].
Ursodeoxycholic acid has been shown in several small studies to reduce gallstone formation post-bariatric surgery. These studies are difficult to compare as they used different doses of ursodeoxycholic acid (i.e. 500 [67] to 1000 [68] mg/ day), for different lengths of time (i.e. 3 [68] to 24 [67] months). The presence of gallstone formation is based upon different diagnostic imaging modalities. Miller [68], in the longest study to date, found that after 24 months the incidence of finding gallstones by ultrasound or CT scan was 8% in 76 patients treated with 250 mg ursodeoxycholic acid bid for six months postoperatively versus 30% in 76 patients treated with placebo. The true incidence of symp tomatic cholecystitis in unknown. Miller [68] reported that 4.7% of patients in the ursodeoxycholic group and 12% in the placebo group underwent postoperative cholecystec-tomy for symptomatic gallstone disease. Though adverse effects of ursodeoxycholic acid are minor (nausea, consti pation), and were not found to be significantly different in the treatment and placebo groups [68], ursodeoxycholic acid is relatively expensive and the pills are large, making passage through a small gastric reservoir difficult. Post operative prophylactic treatment with ursodoxycholic acid at this time is not routinely recommended.
Diverticular disease and diverticulitis
The relationship between diverticular disease and obesity remains unclear. Obese patients are more likely than patients of normal body weight to present with diverticu litis and diverticular perforations. This association has been particularly noted in younger individuals [69, 70], in whom the diagnosis may be missed [71]. In one retrospec tive case series, individuals diagnosed with recurrent diverticulitis or diverticular perforations were significantly more likely to be obese [72]. Obesity has also been identi fied as a significant risk factor for mortality following elec tive sigmoid resection done specifically for diverticular complications.
Cancer
Several mechanisms have been proposed to explain the increased risk of malignancy in obese individuals. These include the insulin and insulin-like growth factor axis, the role of sex steroids and the influence of adipokines. Hyperinsulinemia or insulin resistance reduces insulin-like growth factor (IGF) binding protein, which increases free IGF-1 levels. Both insulin and IGF-1 are associated with increased cell growth and proliferation and both may promote neoplastic cell growth [73]. Hyperinsulinemia also reduces sex hormone-binding globulin, which leads to increased estrogen and androgen levels. These hormones have been shown to be involved in the regulation of cell differentiation, proliferation and apoptosis, and to selec tively enhance the growth of preneoplastic and neoplastic cells [74]. Increased serum leptin, an adipocyte-derived hormone is considered to be an obesity signal, which acts centrally as a catabolic agent to regulate body weight. In vitro leptin has been found to stimulate cancer cells derived from the colon and esophagus [75, 76]. Obesity is also thought to be involved in the immunologic or inflam matory response as described above. Increased levels of cytokines and adipokines, such as adiponectin [77] and transforming growth factor beta [78], may contribute to cell growth and differentiation. A large prospective cohort study assessing cancer mortality in over 900,000 Americans determined that the death rate from all cancers (GI and non-GI) was 52% higher in obese men and 62% higher in obese women compared to men and women of normal body weight. The authors suggested that more than 90,000 deaths per year from cancer could be avoided by maintain ing a BMI less than 25kg/m2 throughout life. The strongest association between obesity and gastrointestinal cancer risk has been demonstrated for esophageal, gall bladder, pancreas, liver and colorectal cancers [79]. A recent system atic review and meta-analysis found GI malignancies to be more prevalent in obese individuals, with a 5kg/m2 increase in BMI leading to an increased risk for gall bladder (RR = 1.59), esophageal (RR = 1.51) and colon adenocarci-nomas (RR = 1.09) in women, and esophageal (RR = 1.52) and colon (RR = 1.24) adenocarcinomas in men [80]. For colon cancer, the association was significantly stronger in men than in women, an observation that gives credence to the finding that estrogen plus progestin may be protective against colon cancer formation [81].
Barrett’s esophagus and esophageal adenocarcinoma
Both GERD and high BMI are considered risk factors for esophageal adenocarinoma. In a recent meta-analysis, indi viduals with a BMI > 25kg/m2 had an OR of 1.52 for esophageal adenocarcinoma compared to an OR of 2.78 in individuals with a BMI > 30kg/m2 [37]. Whether or not this risk is directly mediated by obesity remains unclear. It appears that the risk for Barrett’s esphagus, the precursor to esophageal adenocarcinoma, may be entirely mediated through the increased risk of GERD in obese individuals [82]. Nevertheless, recent studies have suggested that adi-pokines like adiponectin and leptin [83] may be directly associated with Barrett’s metaplasia. Whether or not weight loss results in a decreased risk of esophageal adenocarci-noma is not known.
Proximal gastric adenocarcinoma/cancer of the gastric cardia
The difficulty in endoscopically differentiating proximal gastric adenocarcinoma from distal esophageal adenocar-cinoma complicates risk factor assessment. Several studies have confirmed that obesity is a significant independent risk factor for the development of proximal gastric adeno carcinoma [84–88] but not gastric non-cardiac adenocari-noma [84, 85, 88]. In a large study of over 500,000 US patients, a BMI > 35kg/m2 was found to be a significant risk factor for esophageal and gastric cardia adenocari-noma, with a hazard ratio of 2.27 and 2.46 respectively [84]. The association between these two cancers is unclear, as GERD was found to be a significant risk factor for the development of esophageal adenocarcinoma (see above esophageal adenocarcinoma section) but not for proximal gastric cancers [84, 88].
Pancreatic cancer
The only universally accepted risk factors for the develop ment of pancreatic cancer are smoking and family history [89]. Data regarding any association with obesity and pan-creatc cancer risk are based largely on case-control studies and may be biased by high fatality rates. The RR of obesity as a risk factor for pancreatic cancer was reported to range between 1.2 and 3.0. A recent meta-analysis of six case-control and eight cohort studies found a weak RR of 1.19 for men with a BMI > 30kg/m2 compared to individuals with a BMI < 22kg/m2, but not for women [90]. Not all studies corrected for the significant confounders of smoking and diabetes.
Much like the mechanism by which non-alcoholic stea-tosis leads to liver fibrosis and cirrhosis [91], it has been suggested that non-alcoholic fatty pancreas disease may lead to non-alcoholic steatopancreatitis, which can progress to pancreatic fibrosis and dysfunction (chronic pancreati tis), and eventually pancreatic cancer. The adipose-derived cytokines, IL-1 beta and TNF- alpha are thought to promote this progression [92].
Hepatocellular cancer (HCC)
The relative risk of mortality from primary HCC is 1.68 times higher among women and 4.52 times higher for men, with a baseline BMI ≥ 35kg/m2 compared to a BMI of 18.5–24.9 kg/m2 [79]. In men, HCC had the highest obesity-related relative risk of all cancers. Hepatocellular cancer is a well-recognized complication of cirrhosis, and non-alco holic fatty liver disease associated with obesity and the metabolic syndrome is a known cause of cirrhosis. A study, by Nair et al. [93] of nearly 20,000 US patients found obesity to be an independent risk factor for HCC. However, on multivariate analysis, obesity was only found to be a sig nificant risk factor in patients with either alcoholic or cryp-togenic cirrhosis. This association was not found in patients with other causes of liver cirrhosis such as hepatitis C viral (HCV) infection. Similarly, diabetes mellitus has been sug gested to be a risk factor for HCC development [94, 95], though obesity is a significant confounding variable. A recent study in nearly 1500 patients with chronic HCV cir rhosis followed for a mean of six years found that being overweight or obese were significant independent risk factors for HCC development, with a hazard ratio of 1.86 and 3.10 respectively [96]. Obesity, diabetes mellitus and hyperlipidemia are conditions known to have a synergistic effect on the progression of liver fibrosis in patients infected with HCV [97]. It is difficult to know whether fatty infiltra tion worsens HCV progression itself or whether non-alco holic fatty liver disease is a potential secondary cause of liver cirrhosis in obese individuals.
Cholangiocarcinoma
Studying the risk factors associated with the development of cholangiocarcinoma is hindered by the fact that it is such a rare disease, and that only 10% of cases are associated with a recognized risk factor [98]. Obesity has been sug gested to be a potential risk factor in some studies [99,100] but not in others [101]. As with other cancers, studies are limited by the presence of confounding variables. Specifically, gallstone disease is a recognized risk factor for the development of cholangiocarcinoma, and obesity is a recognized risk factor for the development of gallstones [102].
Colorectal adenocarcinoma
A recent meta-analysis of 31 prospective studies performed between 1966 and 2007 found that a five-unit increase in BMI was associated with a 30% increased risk of colon cancer in men and a 12% increased risk in women [103]. BMI was a significant risk factor for the development of proximal colon cancer in men, and with distal colon cancer in both sexes. Rectal cancer was significantly related to BMI in men but not in women. Abdominal obesity showed a stronger relationship than overall obesity [103]. The reason for the apparent sex difference in the association between colon cancer rates and obesity is unclear. While obesity imposes a greater risk of colon cancer for men of all ages, the risk has been reported to be higher in premenopausal than in postmenopausal women [104]. Although, increased estrogen levels and insulin resistance in premenopausal women have been suggested to contribute [105], this would not explain why colon cancer risk is higher in men than in women [80] or why exogenous estrogen in the form of hormone replacement therapy may be protective [81].
Confounding factors for all studies related to cancer, but specifically to colon cancer, include the influence of exer cise and diet. Physical activity is associated with lower rates of colon cancer [106]. The theoretical explanation for this is that physical activity increases colonic motility [107]. Dietary factors which have been associated with an increased risk of colon cancer include the high consump tion of red meat, low consumption of fresh fruits and veg etables, low-fiber diets and diets low in certain vitamins and minerals including calcium and folate [108]. These diets may be more common in overweight individuals.
Gastrointestinal dysmotility
Alterations in gastrointestinal motility have been recog nized in obese individuals, resulting in a variety of symp toms, most notably reflux, nausea, diarrhea and bloating.
Esophageal dysmotility
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


