Fig. 66.1
Factors associated with the pathogenesis of NAFLD. PNLA pectin lyase, APOC apolipoprotein C, TNF tumor necrosis factor, KLF Kruppel-like factor, PUFA polyunsaturated fatty acids, NAFLD nonalcoholic fatty liver disease, NASH nonalcoholic steatohepatitis
Specific dietary factors either protect against or exacerbate the development and progression of NAFLD. Musso et al. analyzed 7-day diet records from 25 biopsy-proven adults with NAFLD and 25 controls, finding that saturated fat and cholesterol intakes were higher in cases than controls, whilst intakes of polyunsaturated FAs (PUFA), fibre and the antioxidant vitamins C and E were lower [47]. A Japanese study reviewed the 3-day food diaries of 28 adults with NASH and 18 with simple steatosis and identified a significantly lower zinc intake in patients with NASH [48]. Food-based analyses have suggested that higher meat and fructose [49–51], and higher consumption of low-nutrient, high-calorie, and high-salt food [52] are associated with NAFLD. Fructose has been identified as a particular culprit in increasing fat, inflammation and fibrosis [53, 54]. Few pediatric studies of dietary composition have been undertaken in NAFLD; however, a recent large study from Australia compared a Western diet in 993 14-year-olds to later development of NAFLD on US and found a significant association of Western diet and development of steatosis at 17 years [55]. A study in 82 obese Greek children revealed that a diet higher in carbohydrates, saturated FAs and lower in omega-3 was associated with NAFLD [56].
Dietary chemical composition of FAs may be critical factors in lipotoxicity observed in IR. Palmitic acid rather than oleic acid results in lower steatosis but in higher cell death and impaired insulin signaling [57]. A study of fish intake and omega-3 FA intake in children with NAFLD revealed a dietary deficiency of both was associated with increased portal and lobular inflammation [58].
Vos et al. described an association of dietary vitamin E insufficiency and increased steatosis in children with NAFLD [59].
The importance of the intrauterine environment and early infant feeding on development and progression of NAFLD has been a focus of interest. Offspring of obese mice were found to have a dysmetabolic, insulin-resistant and NAFLD phenotype compared to offspring of lean dams [60]. Offspring of lean dams suckled by obese dams developed an exaggerated phenotype suggesting that development of NAFLD and the metabolic syndrome is significantly influenced by early postnatal nutrition. Nobili and colleagues have studied breast-feeding habits in a cohort of children with NAFLD and concluded that breast-feeding is protective against progression of the disease from simple steatosis to steatohepatitis and fibrosis [61]. Figure 66.1 shows the factors associated with the pathogenesis of NAFLD.
Pathophysiology
NAFLD can be thought of as the hepatic manifestation of the metabolic syndrome (linking obesity, IR, hypertension and hyperlipidaemia). The pathogenesis of the condition is still incompletely understood.
IR is found in up to 80 % the children with NAFLD and has a similarly high prevalence in adults with the condition [20, 22, 62, 63]. It is widely accepted that IR and the resulting hyperinsulinemia seem to play a major role in the development of hepatic steatosis and steatohepatitis. The molecular mechanism leading to the involvement of IR in the development of NAFLD is complex, however, and has not yet been fully elucidated.
The ‘two hit hypothesis’ proposed in 1998 consists of a first hit of liver fat accumulation which is caused by an imbalance in uptake and synthesis of hepatic lipids on the one hand and export and oxidation on the other in the context of IR [64]. The steatotic liver is then thought to be more vulnerable to a ‘second hit’. This may be oxidative stress, small-bowel bacterial overgrowth [65, 66], adipocytokines from a high visceral fat mass or saturated free FAs (FFA) [67]. A more recent hypothesis suggests that fat accumulation in the liver is a manifestation of stress and a protective mechanism in itself [68, 69]. This protective effect was demonstrated by Yamaguchi et al. in db/db mice fed a methionine–choline-deficient (MCD) diet [68]. Knockdown of TG synthesis increased the amount of potentially damaging FFA (i.e. blocked detoxification) in the hepatocyte and led to increased necro-inflammation and fibrosis despite decreased steatosis. Thus, in some individuals, the capacity for compensation via detoxification is exceeded and apoptosis and inflammation result.
Steatosis
Macrovesicular steatosis is characterized by the accumulation of TG (formed of glycerol esterified to three FAs) in the hepatocyte. Steatosis is conventionally thought to arise from increased hepatic supply of FFA as a result of obesity and associated extrahepatic IR.
Normally, adipocytes store fat after meals and release fat during fasting by lipolysis. In the liver, carbohydrate is stored as glycogen and when liver is saturated, de novo lipogenesis occurs via acetyl coenzyme A and FA synthetase. The third source of FAs as a substrate for the liver is dietary.
In the setting of normal insulin sensitivity, FAs undergo esterification to TG in the hepatocyte and are then exported from the cell as very low-density lipoproteins (VLDL) via apolipoprotein B enzyme activity [70]. Alternatively, they may undergo beta oxidation in the mitochondria or oxidation in the peroxisomes or microsomes. Uptake of FFA into the mitochondria requires carnitine palmitoyl acyltransferase which is inhibited by insulin and malonyl coenzyme A .
The net retention of lipids is the primary problem in steatosis. The most consistent predisposing factor to hepatic FA accumulation is IR, though other factors may be involved.
In normal physiological circumstances, the role of insulin includes glycogen synthesis, glycolysis and protein and lipid synthesis . In the postprandial state, insulin promotes lipogenesis and suppresses lipolysis and gluconeogenesis. The normal fall in insulin with the fasting state, which is accompanied by an increase in glucagon and catecholamines, mediates glycogenolysis and gluconeogenesis. These processes are accompanied by lipolysis and increased lipid oxidation. Sensitivity to insulin is increased by adiponectin and decreased by TNFα [71] .
In the setting of IR, fat-laden and insulin-resistant adipocytes continue to release glycerol and FFA into the circulation, and deliver increased free FFA to the liver [72–74]. This in itself may then induce hepatic IR [75]. Hyperinsulinemia and hyperglycaemia promote de novo lipogenesis via upregulation of the transcription factors sterol regulatory element-binding protein 1c (SREBP1c) and peroxisome proliferator-activated receptor γ (PPARγ) [72]. In addition, insulin increases malonyl coenzyme A (an intermediate of FA synthesis) and inhibits carnitine palmitoyl transferase thereby inhibiting the passage of long-chain FAs (LCFA) into mitochondria for β-oxidation [76]. SREBP1c can also be upregulated by glucose and saturated fats, whereas polyunsaturated FAs (PUFA) lead to decreased expression [77]. Increased glucose levels also stimulate lipogenesis through the activation of carbohydrate response element-binding protein (ChREBP), a transcription factor activating the expression of key enzymes of glycolysis and lipogenesis [78, 79] .
Hyperinsulinemia also results in decreased TG secretion as VLDL by lowering apolipoprotein B synthesis and stability [80, 81]. Hence, hepatic FFA uptake and lipogenesis outweigh FA oxidation and TG secretion leading to hepatic fat accumulation [82]. In the setting of peripheral IR some hepatic insulin sensitivity may be preserved with continuing de novo lipogenesis as a consequence. This is mixed and thought to be medicated through a functioning insulin receptor substrate 1 (IRS-1) which blocks lipid oxidation, but aberrant IRS-2 serine phosphorylation which fails to suppress gluconeogenesis [70] .
Thus, in summary, the first concept in development of NAFLD is the net increase in circulating free FAs in the setting of IR. The adipocyte continues to release FAs even in the fed state as the insulin-mediated switch-off of lipogenesis is not triggered. The hepatocyte itself continues to produce free FAs via de novo lipogenesis. Activation of transcription factors such as SREBP1c, PPARγ and ChREB occurs in the presence of hyperinsulinemia and hyperglycaemia. Impaired free fatty oxidation and VLDL production ensue, both mechanisms which normally neutralize the toxic effects of free FAs .
Oxidative Stress
Mitochondrial FA oxidation and ketogenesis are increased and the transcription factor PPARα is activated as a result of FA accumulation [83] . This results in reactive oxygen species (ROS) which lead to oxidative stress and lipid peroxidation. Cell membranes are damaged and cytochrome c is released from the mitochondrial intermembrane space. In turn, this leads to an imbalance in the flow of electrons over the respiratory chain (RC) creating over reduction of RC complexes which can react with oxygen to form further ROS [84]. ROS may act as a ‘second hit’ in the development of NASH .
Mitochondrial function has been shown to be impaired in patients with severe steatosis and steatohepatitis [85]. Ultrastructural abnormalities of mitochondria have been demonstrated in patients with NASH [86, 87]. It is not clear if this is a primary or secondary phenomenon however. Mitochondrial abnormalities could be a pre-existing condition enabling the excessive production of ROS in the setting of enhanced FFA β-oxidation [86]. This could explain why for the same amount of obesity, or for the same degree of IR, certain patients just have steatosis, whilst others develop NASH and cirrhosis. Genetic polymorphisms could also at least partially explain this difference in susceptibility as some could favour mitochondrial dysfunction [88]. Alternatively, the overload of the mitochondrial RC, the resulting formation of ROS and subsequent lipid peroxidation products may give rise to mitochondrial damage. There is an inverse correlation of peripheral TNFα levels and measures of IR with RC enzyme levels suggesting that IR and cytokine activity may be important in impairment of the mitochondrial RC [89]. Enhanced ROS formation in the vulnerable steatotic liver subsequently triggers lipid peroxidation and the formation of reactive aldehydes such as 4-hydroxynonenal (4-HNE) and malondialdehyde (MDA). These give rise to further mitochondrial damage and ROS formation, resulting in a vicious cycle [84]. Hepatic stellate cells may be activated by these molecules thus leading to fibrosis [90] .
Cytokines and Inflammation
Much of the progression from simple steatosis to steatohepatitis is characterized by an inflammatory response [91]. It is clear from both rodent and human studies that hepatic steatosis is associated with a state of chronic inflammation [92–94]. More specifically, hepatic steatosis in this context is associated with nuclear factor κB (NFκB) activation. FFA can directly activate the pathway via a lysosomal cathepsin B-dependent mechanism [95], as can mitochondrial and endoplasmic reticulum (ER) stress [93, 96]. NFκB is a sequence-specific transcription factor that functions as a proinflammatory master switch during inflammation. It upregulates the transcription of a wide range of inflammatory mediators including TNFα, IL6 and IL1β. Increased production of inflammatory cytokines by hepatocytes leads to Kupffer cell activation with subsequent inflammatory mediator release and hepatic and systemic IR [97].
Animal studies have shown that translocation of bacteria from the gut to the liver via the mesenteric circulation can activate Kupffer cells (via CD14/Toll-like receptor 4 (TLR4) binding) and induce a local and systemic inflammatory response [98]. There has been a great deal of interest in gut microbiota and the innate immune response in the context of obesity and IR [36]. There is evidence that intestinal bacterial overgrowth exacerbates NAFLD and that the prevalence of bacterial overgrowth is higher in those who are obese [37], the portal circulation providing a direct route from gut to liver. Manipulation of gut microbiota and elimination of intestinal bacterial overgrowth may thus be a promising way to halt the progression of steatosis to steatohepatitis and fibrosis.
Finally, visceral fat is a highly inflammatory tissue and the source of many inflammatory mediators known as adipocytokines which have an important role in IR and, most likely, in NAFLD [99, 100]. These adipocytokines, including leptin, adiponectin, TNFα and IL6, are polypeptides produced by both adipocytes and macrophages which infiltrate adipose tissue [101]. Adipokines are involved in the various injury patterns in NASH such as cell death, inflammation and fibrosis [84].
Leptin is a 16-kDa protein, a product of the ob gene and has important roles in appetite suppression and regulation of energy metabolism [102], with high levels in obese individuals though this is thought to be a result of leptin resistance. The role of leptin in NAFLD is not yet clear though it is thought to contribute as a proinflammatory, profibrogenic mediator [103, 104].
Adiponectin is a polypeptide adipokine with a collagen-like domain and globular domain produced in white adipose tissue. It has an important role in insulin sensitivity, as part of their action, thiazolidinediones (TZDs) are known to increase levels of adiponectin [105]. It is also hepatoprotective with anti-inflammatory and anti-fibrogenic properties [106–108].
Hepatocyte Apoptosis
Hepatocyte apoptosis is recognized as an important event in the development of chronic liver disease and has particular prominence in NAFLD [109]. The initiating event in apoptosis may be extrinsically mediated hepatocyte injury (e.g. in autoimmune liver disease, viral hepatitis and ischaemia perfusion injury). This is usually directed though pathways involving Fas ligand, TNFα, and TNF-related apoptosis-inducing ligand (TRAIL). Alternatively, intrinsic injury and death may occur via organelle dysfunction when cells are subjected to excessive oxidative stress (ER or mitochondrial), for example, with drugs/toxins, FAs, and iron. This results in altered membrane permeability and RNA damage with cytochrome c release [110, 110]. The injurious mechanism in NAFLD/NASH appears to be due to a combination of extrinsic and intrinsic insults [111, 112].
Though apoptosis is classically thought to be a silent event without provoking an inflammatory response, this is not the case in the liver [111]. Apoptotic bodies can activate stellate cells and Kupffer cells inducing an inflammatory response and leading to the progression of steatohepatitis and fibrosis [113, 114].
Fibrosis
The final common pathway of inflammation , oxidative stress and hepatocellular damage is the development and progression of fibrosis in NAFLD. The process of fibrosis involves the deposition of extracellular matrix within the parenchyma. Cirrhosis, the end stage of the fibrotic process, is characterized by septum and nodule formation. Several different injurious processes will result in fibrosis. Hepatocyte injury, inflammation, apoptosis and death initiates the process which involves a cascade of inflammatory cells, the release of cytokines and the activation of fibrogenic effector cells (mainly stellate cells) [115].
Thus, a number of different processes and mechanisms are involved in the progression of steatosis to NASH: oxidative stress, inflammation , apoptosis and fibrosis. The exact sequence of development of obesity , fatty liver and NAFLD remains unclear. Whether IR causes hepatic steatosis or whether the accumulation of fat in the liver is the primary event leading to hepatic and peripheral IR is also yet to be elucidated [116]. Figure 66.2 shows the interplay of oxidative stress, inflammation, apoptosis and fibrosis in the pathogenesis of NASH [117].
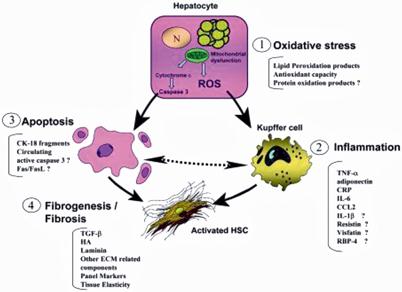
Fig. 66.2
The interplay of oxidative stress, inflammation, apoptosis and fibrosis in the pathogenesis of NASH. (ROS reactive oxidative species, HSC hepatic stellate cell. Reprinted with permission from [117], with permission from John Wiley and Sons)
Diagnosis and Histology
Children with NAFLD are often asymptomatic or may present with vague nonspecific symptoms such as abdominal pain and/or fatigue. The majority are overweight (gender- and age-specific BMI > 85th percentile) or obese (> 95th percentile) [118]. Hepatomegaly may be present and acanthosis nigricans (a black pigmentation of the skin folds, axillae and neck), often seen in children with IR, is found in 30–50 % of children with NAFLD [20, 119]. The majority of children with NAFLD have IR as measured by homeostasis model assessment-IR (HOMA-IR is (fasting glucose (mmol/l) × fasting insulin (IU/l))/22.5) [20]. A normal HOMA-IR (< 90th percentile) is generally less than 2.5 but varies with sex and pubertal stage [120]. Children will often have a positive family history for the metabolic syndrome [19].
In the diagnostic workup of NAFLD, alternative causes of chronic liver disease should be excluded including chronic hepatitis B and C infection, Wilson disease, α1-antitrypsin deficiency, autoimmune hepatitis and drug toxicity (including steroids, amiodarone, oestrogens and antiretroviral treatment). Conditions such as cystic fibrosis, malnutrition and parenteral nutrition-associated liver disease may also present with a fatty liver on US and can be excluded on clinical or biochemical grounds. In addition, mitochondrial/ metabolic disease and cholesterol ester storage disease may also look very similar on liver biopsy and need to be considered. Table 66.1 gives differential diagnoses that must be excluded.
Table 66.1
Conditions which need to be excluded before a diagnosis of NAFLD is made
Clinical condition | Clinical features | Biochemical/ other features which help distinguish from NAFLD |
|---|---|---|
Wilson disease | May present with chronic liver disease, hemolytic anaemia or more rarely neurological disease in childhood. Look for Kaiser Fleisher rings | Low serum ceruloplasmin, high urinary copper pre and post penicillamine, high liver copper, genetics may be positive |
Alpha 1 antitrypsin deficiency | – | Alpha 1 anti-trypsin phenotype ZZ (or ZS) more rarely SS |
Drugs—steroids, amiodarone, alcohol, methotrexate, ecstacy, l-asparaginase, vitamin E, valproate, tamoxifen, antiretrovirals | History of drug ingestion | – |
Cystic Fibrosis-associated liver disease | History/examination | Positive sweat test/genetics positive |
Malnutrition | Clinical examination | – |
Coeliac disease | May have failure to thrive | Tissue transglutaminase positive/positive jejuna biopsies |
Hepatitis C | – | Positive serology |
Parenteral nutrition-associated liver disease | Compatible history | – |
Mitochondrial disease/fatty acid oxidase deficiency | May be history of neuro-developmental problems or other system involvement | Abnormal respiratory chain enzymes liver/muscle, abnormal acyl-carnitines,/skin fibroblast studies, mitochondrial depletion demonstrated, genetics for mitochondrial disease |
Metabolic disease: Lysosomal acid lipase deficiency (cholesterol ester storage disease) | – | Positive enzymology or genetics |
Galactosaemia | Age of presentation | Abnormal Gal-1-PUT result |
Fructosemia | May be history of avoiding sweets | Enzymology on liver biopsy |
Glycogen storage disease | Hepatomegaly, may be short, history of fasting hypoglycaemia | Positive enzymology/genetics, glycogen not fat on biopsy |
Peroxisomal disorders | May be hypotonic/have wide anterior fontanelle, neurological problems | Abnormal very long-chain fatty acids |
Mauriac syndrome | History type 1 diabetes | Glycogen on liver biopsy |
Hypobetalipoproteinaemia/abetalipoproteinaemia | Low serum triglycerides, may be history of fat malabsorption, may be failure to thrive | Low or absent Apo1B levels |
Lipodystrophies | Examination | Genetics |
Schwachman syndrome | FTT pancreatic insufficiency, bony changes, cyclical neutropenia | Genetics |
Recommendations for undertaking biopsy in children with suspected NAFLD are made by a consensus document published by the American Association for the Study of Liver Diseases (AASLD), American College of Cardiology (ACG) and American Gastroenterological Association (AGA) [121]. These are to perform liver biopsy where the diagnosis is unclear there is a possibility of multiple diagnoses, before starting therapy with potentially hepatotoxic medications or prior to starting children on pharmacologic therapy for NASH. An ESPGHAN guideline was published the same year suggesting that liver biopsy should be performed in children with suspected NASH according to the following criteria; to exclude other treatable disease, in cases of clinically suspected advanced disease, as part of an interventional protocol or research trial [122]. Table 66.1 shows conditions which need to be excluded before a diagnosis of NAFLD is made.
Tools commonly used in the workup and diagnosis of NAFLD include serum transaminases and imaging techniques (US, computerized tomography, magnetic resonance imaging/spectroscopy). The sensitivity, specificity and predictive values of these techniques are variable [3]. Franzese et al. studied the incidence of liver involvement in 72 obese children using both US and transaminases [123]. Fifty-three percent of the children had a bright liver on US consistent with liver steatosis whilst only 25 % had elevated transaminases. Molleston and colleagues reported histological abnormalities in 91 children with NAFLD and normal or only mildly elevated ALT. Forty-six percent of the group had some fibrosis with 38 % mild to moderate fibrosis and 8 % bridging fibrosis [124]. Neither US nor transaminases are good discriminators of histological severity [125]. Ultrasound will detect > 20–30 % steatosis as increased echogenicity, though this is not specific for fat [126]. Magnetic resonance imaging/spectroscopy (MRI/MRS) is more sensitive and can detect > 5 % steatosis [127]. However, neither technique can assess presence of inflammation or fibrosis.
Liver biopsy remains the gold standard in differentiation of steatosis from steatohepatitis. The diagnosis of NASH is based on a specific pattern of histopathological findings including macrovesicular steatosis, mixed or polymorphonuclear lobular inflammation, ballooning degeneration with Mallory hyaline, a perivenular distribution of fibrosis in adults (type 1 NASH) [128] (Fig. 66.3). Children may have a different pattern of disease with greater degree of steatosis, less prominent ballooning and portal rather than pericentral accentuation of inflammation and fibrosis (type 2 NASH) [129] (Fig. 66.4). Schwimmer et al. reviewed the histological findings in a cohort of 100 children (2–18 years) with biopsy-proven NAFLD [125]. Type 1 NASH was present in 17 % and type 2 NASH in 51 % of the children. Sixteen percent of biopsies had overlapping features of type 1 and 2 disease and the remaining 16 % showed simple steatosis. Children with type 2 NASH were younger and had greater severity of obesity than in type 1 NASH. Boys and those of Asian, Native American and Hispanic ethnicity were more likely to have type 2 NASH. In contrast, in a study from Italy of 57 children with NASH, only 2.4 % had type 1 NASH, 28.6 % were classified as type 2 NASH, whereas the majority (52.4 %) had an overlap between the two (17 % had simple steatosis) [130]. Takahashi reports the presence of type 2 NASH in 9 % of adult patients and 21 % of pediatric patients studied in Japan [131]. The mechanism leading to the different phenotypes of NAFLD is not yet understood.
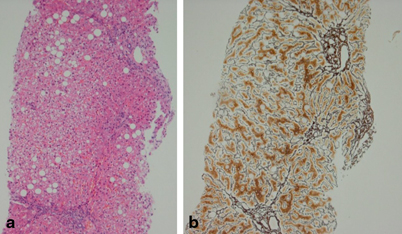
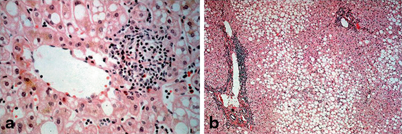
Fig. 66.3
Histology of NAFLD. a Hematoxylin and eosin staining showing steatosis, inflammation and ballooning. b Reticulin stain showing fibrosis Pictures kindly provided by Department of Liver Histopathology, King’s College Hospital
Fig. 66.4
Liver biopsies showing a type 1 NASH with pericentral disease and b type 2 NASH with periportal disease. Type 2 NASH is more common in children and type 1 in adults. Pictures kindly provided by Department of Liver Histopathology, King’s College Hospital
The occurrence of portal inflammation was reviewed as a distinct entity in NASH by the NASH Clinical Research Network (CRN) [132]. A study of biopsies from 728 adults and 205 children found that the presence of portal inflammation in adults was associated with older, female patients with a higher BMI and IR. There was a clear association with amount and location of steatosis, ballooning and advanced fibrosis. In the pediatric group, portal inflammation was associated with younger age, azonal location of steatosis and more advance fibrosis (bridging). In both groups, it was associated with diagnosis of definitive NASH. There was no association with lobular inflammation in either group. It is not clear if this pattern is due to a separate pathophysiological mechanism, though it certainly seems to be a marker of more advanced NASH. The periportal pattern mirrors that of the ductular reaction which has been reported in NAFLD. The possible epithelial-mesenchymal transition of biliary cells in this process may relate to the pattern of fibrosis seen [133].
Though the classic description of fat in NAFLD is macrovesicular, the presence of microvesicular steatosis has recently been described in 102 of 1022 biopsies in patients with the condition [134]. In this study, the presence of microvesicular steatosis was strongly associated with cellular injury and cytoskeletal damage. Microvesicular steatosis has the appearance of distended hepatocytes with foamy cytoplasm; the nucleus is usually central rather than pushed peripherally as in macrovesicular steatosis. Oil red O staining is sometimes needed to identify microvesicular steatosis if it is not visible in haematoxylin and eosin staining. Classically, this type of steatosis has been associated with mitochondrial disease, acute fatty liver of pregnancy and some drug effects (e.g. steroids and valproate) which can cause β-oxidation impairment [135]. Taken together, microvesicular steatosis in NAFLD is a likely indicator of mitochondrial damage. It is not yet understood if this is a feature of advanced disease per se or if the pathogenesis of disease in those with microvesicular steatosis is different.
Caldwell et al. reported on the significance of ballooning in NAFLD [136]. These are classically enlarged cells with rarefied cytoplasm. Using ultrastructural analysis, this group reported on the multiple small fat lipid droplets seen in with degree of ER dilatation and Mallory Denk bodies and cytoskeletal disarray. Fugii et al. have demonstrated altered expression of FFA-associated protein on the surface of fat droplets which also stain for oxidized phosphatidylcholine (a marker of oxidative damage) [137]. They concluded that oxidative injury to the fat droplet surface may impair its safe disposal and contribute to lipotoxicity.
The Pathology Committee of the NASH CRN proposed a histological scoring system that could be useful in studies of NAFLD [138]. The scoring system includes the evaluation of steatosis (0–3), lobular inflammation (0–2), hepatocellular ballooning (0–2) and fibrosis (0–4). The NAFLD activity score (NAS) is the unweighted sum of steatosis, lobular inflammation and hepatocellular ballooning scores. NAS of 5 or more correlates with the diagnosis of NASH, whilst NAS less than 3 is defined as ‘not NASH’. As this system is typically developed for adult type 1 NASH, the interobserver agreement for pediatric NASH is not as strong (only 18 children were included in the study cohort used for development of the score). The CRN also emphasize that the scoring system was developed as a tool for use in trials and is not a surrogate for a histological diagnosis of NASH [139]. Despite these shortcomings, this is the best available tool to standardize the description of the entire spectrum of NAFLD in both adults and children across different centres for research purposes.
It is important, however, to consider the pediatric pattern of disease as a separate entity, particularly when investigating the pathophysiological mechanisms or putative biomarkers of disease severity/progression. Figure 66.3a and b shows an example of histology of NAFLD. Figure 66.4 shows showing type 1 NASH with pericentral disease (a) and type 2 NASH with more periportal disease (b).
Natural History and Management
The natural history of NAFLD varies according to the histological pattern of the disease. Simple steatosis without evidence of inflammation or fibrosis appears to have a benign course whilst NASH is a potentially serious condition which can progress to cirrhosis. In a 10-year follow-up of 132 individuals with NAFLD, 22 % of those with NASH versus 4 % of those with simple steatosis went onto develop cirrhosis [46]. There was also a significantly higher liver-related mortality in the time period in the NASH group. Ekstead et al. followed 129 patients over a mean period of 13.7 years and found progression of liver fibrosis in 44 % [140]. All-cause mortality was higher in the NASH group versus those with simple steatosis, particularly from cardiovascular and liver-related deaths. Four hundred and twenty patients with NAFLD were followed in the Rochester Epidemiology project, a population-based study. Liver-related mortality was the third leading cause of death in those with NAFLD [6]. Cirrhosis was present at follow-up in 5 % of cohort. Adams et al. reported a series of 103 patients with NAFLD with follow-up biopsy at a mean interval of 3.2 years and found that fibrosis progressed in 37 %, remained stable in 34 % and regressed in 29 % [141]. A higher BMI and diabetes were associated with fibrosis progression. A systematic review of 10 studies including 221 patients with a mean follow-up of 5.6 years concluded that fibrosis progressed in 37.6 % of those with NASH [142]. Musso reported a further systematic review of 40 cohort studies examining the natural history of NAFLD in adults [143]. The authors found an overall and cause-specific pooled increase in mortality in those with NAFLD (odds ratio (OR) 1.57 (1.18–2.1)) versus the general population. In those with a steatotic liver on US, the risk of mortality from cardiovascular disease was increased by an OR of 2.05, with an OR of 3.5 for the development of type 2 diabetes. With subgroup analysis, patients with simple steatosis were found to have a similar mortality as the general population and those with NASH had an OR of 1.81 for overall mortality. The excess cause of death in this subgroup was mainly liver related with an incidence of 11–17 versus 1.7–2.7 % in those with simple steatosis.
Steatosis is often no longer present in end-stage NASH and is replaced by cirrhotic change. NAFLD is thought to be the underlying etiology in the majority of cases of cryptogenic cirrhosis in many parts of the world [144, 145]. Evidence of this includes the finding that the prevalence of obesity and type 2 diabetes in cryptogenic cirrhosis mirrors that of those with NAFLD [146]. There is also an increased frequency of NASH occurrence post transplant in those with cryptogenic cirrhosis [147].
The development of HCC remains a major concern in patients with NAFLD [148]. Multiple case series and reviews have reported the association of HCC with the condition, even in the absence of NASH-related cirrhosis [149–154]. Diabetes and obesity are known to be risk factors for HCC and other malignancies [150]. In the setting of NASH the inflammatory cascade, oxidative stress and IR may all contribute to hepatocyte hyperplasia [150].
Cirrhosis secondary to NASH has been reported in children as young as 10 years [119, 155]. A recent study by Feldstein et al. describes the long-term outcome of 66 children with NAFLD followed-up for up to 20 years [156]. Of the five children who underwent follow-up biopsy, four showed progression of fibrosis. During the study period, two patients required liver transplantation for decompensated end-stage disease. Both had recurrence of NASH in the allograft and one required retransplantation.
In adult studies, the variables most commonly associated with fibrosis are the presence of diabetes, increasing age and high BMI [157]. Similarly in children, severity of obesity and IR seem to be predictors of advanced fibrosis [20]. The difference between the natural history of type 1 and type 2 NASH has not yet been characterized and is an important subject for future research.
Management of NAFLD encompasses lifestyle modification, medication or both. NAFLD is largely the consequence of imbalanced nutrition and sedentary behaviour on the background of genetic predisposition. Primary prevention is the ideal. In adults, weight reduction of 5–10 % body weight often leads to normalization or improvement of serum transaminases and reduced hepatic steatosis, inflammation and fibrosis [158–160]. In children, weight maintenance as the child crosses the height percentiles may achieve the same effect.
Several case series and uncontrolled trials have demonstrated the effect of weight loss on improvement of transaminases or US abnormalities [123, 161, 162]. A prospective study carried out in 84 children (3–18.8 years) with NAFLD demonstrated a significant decrease in BMI, levels of fasting glucose, insulin, lipids, transaminases and liver echogenicity on US following a 12-month program of lifestyle advice consisting of diet and physical exercise [130]. Another study of 53 children comparing lifestyle intervention plus antioxidant or lifestyle intervention plus placebo demonstrated similar improvements in both groups in terms of steatosis, inflammation, ballooning and NAS score [163].
Control of both quality and quantity of dietary components may be important. As above, nutritional data to date have suggested that a high intake of simple carbohydrates such as fructose with a low intake of polyunsaturated FAs correlate with pathogenesis and progression of disease [47, 53].
The type of fat consumed is possibly more relevant than quantity with higher saturated fat and lower PUFA intake associated with IR and NAFLD in some studies [47, 52]. Palmitic acid rather than oleic acid results in lower steatosis but in higher cell death and impaired insulin signalling in vitro [57]. Several small studies of PUFAs in adults and one in children have demonstrated improved liver enzymes and histology in the treatment group [164–167].
Insulin Sensitizers
Insulin sensitizers have been investigated extensively and in view of the strong association of IR and the progression of NAFLD. Metformin is an important insulin sensitizer, reducing hyperglycaemia through a number of different mechanisms including inhibition of hepatic output and increasing peripheral uptake [168]. Thiazolidiones are PPARγ agonists which stimulate FFA uptake by adipocytes and allow the redistribution of fat from the liver to peripheral tissue [105].
A Cochrane review examined the evidence for insulin sensitizers in NAFLD. Only three randomised controlled trials (RCT) met search criteria (RCT comparing insulin sensitizer to placebo in nondiabetic adults with NAFLD) [169]. Though there was some histological and biochemical improvement with both metformin and rosiglitazone versus placebo, the results of the meta-analysis are inconclusive [170–174]. A large RCT (the PIVENS (pioglitazone, vitamin E or placebo for the treatment of NAFLD) trial) found favourable results for vitamin E but not for pioglitazone [175].
Metformin has the side effect of gastrointestinal intolerance but weight loss is an advantage of this medication. The TZDs have the unfortunate side effect of weight gain. There have also been concerns about cardiovascular events and diminished bone mass with their use [169]. There is no available data on the safe use of TZDs in children.
Only metformin has been investigated in pediatric patients with NAFLD. A large randomized, double-blind, placebo controlled trial has recently been reported by the NASH CRN in which both vitamin E and metformin were used [176]. The purpose of this study was to determine if therapeutic modification of IR or oxidative stress leads to improvement in serum or histological indicators of liver injury and quality of life (treatment of nonalcoholic fatty liver disease in children (TONIC) trial). The results demonstrate that there was a significant histological improvement in the vitamin E treatment group versus placebo with resolution of NASH ( p < 0.006), though there was not a significant difference in resolution of transaminases (the primary outcome measure) in either treatment group. Other studies of metformin in children include an open-label pilot study of metformin (500 mg twice daily for 24 weeks) which was conducted in 10 nondiabetic children with biopsy-proven NASH and elevated ALT level [177]. Significant improvement was observed in serum ALT and hepatic steatosis as assessed with MR spectroscopy. A subsequent study conducted in children did not show any benefit of Metformin compared to lifestyle advice [178]. A third study of metformin in insulin-resistant adolescents resulted in lower severity scores of fatty liver on US and a decrease in prevalence of fatty liver disease in the metformin group [179].
Antioxidants
The oxidation of FFA leads to the production of reactive oxygen species. Normally, hepatocytes have enough antioxidants such as glutathionine to keep this process under control. The increased oxidative stress in NASH is in part due to increased FFA which overwhelm a presumably impaired normal oxidation pathway leading to the abundant production of reactive oxygen species. As this may have a major pathophysiological role in development and progression of the disease, antioxidants and hepato-protectants may be useful in management of the condition. Antioxidants such as ursodeoxycholic acid, vitamin E, S-adenosylmethionine, betaine, N-acetyl cysteine and pentoxyfyline have all been investigated in this context. Newer drugs such as caspase inhibitors and inhibitor of kappa B kinase (IKK) inhibitors are under development. The TONIC trial demonstrated some encouraging histological improvement with vitamin E; however, there was no difference to outcome measures as defined [180].
An open-label pilot study with vitamin E (400–1200 IU/day) in 11 children with NASH, showed improvement of serum transaminases despite no major changes in BMI or US appearance of the liver [181]. Other studies using vitamin E in children refute these findings [178, 182].
The recent PIVENS trial demonstrated that in a multicentre, double-blind, RCT of vitamin E versus pioglitazone versus placebo in 248 nondiabetic adults with NAFLD, there was a significant improvement in histology in the vitamin E group (43 %) versus the pioglitazone group (19 %) [175].
Ursodeoxycholic acid (UDCA) has multiple cytoprotective properties [183]. Using a combination of UDCA and vitamin E, Balmer et al. demonstrated an improvement in transaminases and liver histology together with increased adiponectin levels and decreased hepatocyte apoptosis [184]. There was no significant change in the amount of fibrosis present following 96 weeks of treatment, however. UDCA has also been studied in pediatric NAFLD. A small study which evaluated the efficacy of UDCA in 31 obese children with abnormal transaminases did not show any effect of UDCA treatment [185]. A Cochrane review found insufficient evidence that UDCA was an effective treatment for NAFLD [186].
Bariatric Surgery
The role of bariatric surgery (both Roux-en-Y bypass and gastric band) in management of NAFLD was examined in adults as a Cochrane review. Though RCT exist to date, 21 observational studies were included. The majority of these studies demonstrated improvement in steatosis or inflammation post surgery though with improvement in fibrosis in only 6 studies and deterioration in 4 [187]. No data are available for children.
Orlistat
Orlistat is a gastrointestinal lipase inhibitor which has beneficial effects in terms of weight reduction and reduction of free FAs. The effect of Orlistat with calorie-controlled diet on liver enzymes and histological features of NAFLD has been tested in three RCTs. These RCTs had conflicting results, and though Orlistat appears to be a safe adjunct to dietary counselling, the net effect is uncertain [188–190]. No pediatric data are available.
Probiotics
There has been recent interest in the role that gut microbiota play in obesity, IR and NAFLD [36]. There is evidence that intestinal bacterial overgrowth exacerbates NAFLD and that the prevalence of bacterial overgrowth is higher in those who are obese [37]. Manipulation of gut microbiota with probiotics is a promising way to halt the progression of steatosis to steatohepatitis and fibrosis. Both animal experiments and small human studies have investigated the use of probiotic agents to reverse the progression of steatohepatitis and fibrosis in NAFLD [191, 192]. Two recent double-blind RCTs—one in adults and one in children were reported [193, 194]. Treatment with probiotics in both studies resulted in improvement in transaminases.
Fibrates
Fibrates are PPARα ligands which can decrease TG levels and increase high-density lipoprotein (HDL) by stimulating lipoprotein lipase and regulating apolipoprotein [195]. Fibrates have been shown to ameliorate IR and inflammation, stimulate β-oxidation and improve transaminase levels but not histology in patients with NAFLD [196]. There have been no RCT in children.
Noninvasive Biomarkers in NAFLD
Though the criterion standard for diagnosis and assessing progression of disease is liver histology , the decision ‘if or when’ to perform a liver biopsy in children with suspected NAFLD remains controversial. Liver biopsy in children requires admission to hospital and sedation. Risks include bleeding and very rarely death [197]. Repeated biopsy is not a suitable tool for regularly monitoring progression of disease or response to treatment. In addition, biopsy samples only 1/50,000 of the liver, raising the possibility of sampling error [198].
There has been much focus on the development and validation of noninvasive biomarkers of NAFLD in recent years. There is an urgent need for a less invasive method than biopsy of screening the population, stratifying disease severity and following disease progression.
The pathophysiology and evolution of the condition under scrutiny is an important consideration in the development and evaluation of biomarkers. In the case of NAFLD, there are two potential targets. The first is the differentiation of simple steatosis from steatohepatitis. This is important as the prognosis of those with simple steatosis is different from those with NASH [199]. The second issue is the identification of fibrosis stage. This is the main determinant of prognosis and knowing the extent of fibrosis is useful in making treatment decisions, in patient selection for treatment studies and in monitoring progression/regression. Most longitudinal cohort studies in NAFLD have shown that prognosis is determined by stage and rate of progression of fibrosis rather than the presence of necro-inflammation [46, 140, 200]. Clinical importance lies with being able to differentiate between no/minimal fibrosis (F0/F1), significant fibrosis (F2), severe fibrosis (F3) and cirrhosis (F4).
Serum Biomarkers and NASH
Large adult series have suggested scoring systems using age, BMI, IR, AST/ALT, platelet count and albumin to differentiate mild from severe disease [157, 201–203]. These simple markers are neither sensitive nor specific enough in isolation [5, 12]. A growing understanding of the pathophysiology of the disease has allowed the investigation of more specific, mechanism-based biomarkers [117, 204, 205].
Markers of apoptosis/cell death have been shown to be very useful in differentiating simple steatosis from NASH [109] . CK18-M30 fragments have been shown by a number of studies including pediatric studies to correlate well with severity of NASH [206–209].
A number of predictive models to differentiate either NAFLD from obese controls or simple steatosis from NASH have been developed and validated. Tools include the hypertension, ALT, IR (HAIR) score which gives an area under the curve (AUROC) of 0.9 [210], and the NASH test (consisting of 13 variables including weight, TG, glucose, α2-macroglobulin and apolipoprotein A) which has an AUROC of 0.79 for differentiation of NASH from simple steatosis [211]. When the NASH test is combined with the Steatotest (10 variables including simple blood tests, age, gender and BMI) [212] and the Fibrotest into what is known as the Fibromax panel, the diagnostic accuracy improves further [212].
The following studies report predictors of NAFLD using routine clinical parameters in cohorts of obese children. Sartorio et al. reported a multivariate analysis of 267 obese children and found that BMI z score, ALT, uric acid, glucose and insulin were useful predictors of NAFLD [17]. Mandato reported IR, ferritin, CRP and glutathione peroxidase as good discriminators of those with NAFLD from those without in a cohort of obese children [205]. Neither of these studies used a histological diagnosis of NAFLD.
Noninvasive Markers of Fibrosis in NAFLD
Simple tests derived from regression analysis of large series of patients include the AST to platelet ratio index [220], the AST to ALT ratio [221], which have been validated in the NAFLD population with AUROC between 0.67 and 0.86 for differentiation of severity of fibrosis [222–224]. Algorithms specifically derived from NAFLD cohorts include the BAAT score (consisting of BMI, ALT, age and TG levels) [225], the BARD score (BMI, AST/ALT ratio, diabetes) [202, 226] and the NAFLD fibrosis score (incorporating age, glucose, AST, ALT, BMI, platelets and albumin) [201, 222, 223, 227, 228]. In a recent meta-analysis the AUROC for the NAFLD fibrosis score was found to be 0.85 with a pooled sensitivity of 90 % and specificity of 97 % [143].
Fibrometer has been validated in a NAFLD population [224]. The test incorporates age, weight, fasting glucose, AST, ALT, ferritin and platelets and demonstrates an AUROC of 0.94 for significant fibrosis, 0.9 for severe fibrosis and 0.9 for cirrhosis.
Other biomarkers measure the degree of ECM turnover. Using such ECM markers is a more direct method of assessing fibrogenic activity, and it will tend to measure a dynamic process rather than a static one.
Combinations of both clinical markers and ECM turnover include the Fibrotest [229, 230] which is an algorithm of 13 markers derived from regression analysis including haptoglobin, α2-macroglobulin, apolipoprotein A1, bilirubin, γGT, age and gender. It has an AUROC of 0.84 for advanced fibrosis in NAFLD [231].
The European liver fibrosis (ELF) test combining hyaluronic acid, procollagen III N-terminal peptide (P3NP) and TIMP1 was first derived by Rosenberg et al. in a cohort of over 1000 patients with chronic liver disease including NAFLD [232] and has since been validated in other NAFLD cohorts with the addition of several simple markers to improve accuracy [233]. Importantly, this test has been shown to correlate well with outcome [234]. In 112 children with NAFLD, ELF had an AUROC of 0.92, 0.98 and 0.99 in distinguishing any, significant and advanced fibrosis respectively [235].
As with adult studies, the noninvasive diagnosis of fibrosis (rather than necro-inflammatory change) in NAFLD is considered separately in children. It is important to acknowledge that the different distribution of fibrosis in pediatric patients may affect the validity of applying measures derived from adult cohorts to this population.
Iacobellis et al. reported a cohort of 69 children with NAFLD, 60 % of whom had fibrosis [203]. They found that BMI was the only significant predictor of fibrosis with multivariable analysis of simple clinial parameters. BMI had an OR of 5.85 for predicting presence of fibrosis. Manco et al. found waist circumference as a significant predictor of fibrosis in a cohort of 197 children with NAFLD (OR 2.4 (1.04–5.54)) [236]. In both these studies the number of children in the F2–F4 groups was small.
Nobili et al. developed and internally validated the paediatiric NAFLD fibrosis index (PNFI) in 136 children with NAFLD [237]. Logistic regression analysis of gender, age, BMI, waist circumference, ALT, AST, γGT, albumin, prothrombin time, glucose, insulin, cholesterol and triglcyerides gave a predictive model with an AUROC for detection of fibrosis was 0.85. Again this study was limited in view of small numbers in fibrosis groups F2–F4.
Hyaluronic acid (HA) level > 1200 ng/ml in 100 children (65 % with fibrosis) was a reliable discriminant of fibrosis in NAFLD in one study [203]. The ELF test was evaluated by Nobili et al. in 122 children with NAFLD [238]. Simple markers including age, waist circumference and TG were added to improve diagnostic accuracy. Excellent AUROC for any (0.92), significant (0.98) and advanced (0.99) disease were achieved. In this cohort, 37 (30 %) had no fibrosis, 58 (48 %) scored as F1, 9 (7 %) as F2, and 8 (6.5 %) as F3–F4. Alkhouri et al. developed this further and validated both the PNFI and ELF in a cohort of 111 children with NAFLD (69 % with fibrosis) [239]. The area under the curve for presence of fibrosis was 0.76 for PNFI, 0.92 for ELF and when the two indices were combined: 0.94. The major issue in both studies was the skew towards no or minimal disease, potentially overestimating the accuracy of the test.
Noninvasive Biomarkers and Imaging
US, CT and MRI
US has a high sensitivity and specificity for diagnosis of steatosis > 30 %, but is not good at detecting fibrosis. Because of the low cost, the absence of radiation exposure and the wide availability, US is often used in screening for NAFLD. The accumulation of fat causes the liver to appear hyperechoic compared with the kidney. This finding is nonspecific and does not differentiate fat from other substances such as glycogen. When compared with histological findings, the sensitivity of US to detect fat infiltration below 30 % of the liver is low [240]. Computed tomography (CT) is rarely used for the assessment of NAFLD in children because of its ionizing radiation exposure. Magnetic resonance imaging (MRI) and spectroscopy are the imaging techniques with the greatest accuracy to determine hepatic fat content in studies of both adults and children [127, 241–243]. Aside from liver fat, however, other features of NASH cannot be assessed. Other methods include MR elastography which visualizes and measures propagating shear waves and has a high sensitivity (> 85 %) and specificity (> 90 %) for fibrosis [244]. Cost of this technique may be preclusive however.
For diagnosis of NASH , Iijima et al. have reported on the use of contrast US with Levovist with an AUC of 1.0 [245]. The decreased accumulation of micro-bubbles with advancing degree of fibrosis is unique to NAFLD.
Transient Elastography
Transient elastography (Fibroscan®) has been shown to be a useful method for detection of liver fibrosis. In NAFLD, a small number of studies have demonstrated the efficacy of TE in distinguishing severity of fibrosis. In a study of 246 adults with NAFLD, TE had an AUROC of 0.84, 0.93 and 0.95 in distinguishing significant fibrosis, severe fibrosis and cirrhosis, respectively [248]. A Japanese study demonstrated similar results [249]. A recent report of 52 children with NAFLD has shown an AUROC of 0.977, 0.992 and 1 for distinguishing any, significant and severe fibrosis [250]. Feasibility and reproducibility of transient elastography is an issue when patients have a BMI > 30 [251]. An XL probe is now available for better accuracy in this scenario [252].
Non-hypothesis Driven Search for Novel Biomarkers Using New Technologies
Future Areas for Research
The pathophysiology of NAFLD in both adults and children is still incompletely understood. The reason for different patterns of disease in children, particularly with reference to the occurrence of periportal inflammation and fibrosis rather than the typical type 1 pattern of pericentral disease remains elusive. Determining susceptibility to the disease using genetic analysis for SNPs may become standard practice, particularly in screening the high number of overweight and obese pediatric patients. Further investigation into specific dietary patterns in children may also yield valuable information in this multifactorial disease.
The determination of the most effective management of the condition—both in terms of achieving lifestyle change and pharmacological treatments—will be a major focus going forward. Further areas of interest include the role of intestinal microbiota and the possible use of probiotics in the condition and dietary manipulation with PUFA.
Finally, the long-term outcome of children with NAFLD remains unknown and well designed, long-term prospective studies using networks, such as the NASH clinical research network in the US, are vital to achieving this.
In conclusion, NAFLD in children is a very real, very prevalent condition and, should current trends continue, is likely to become the most common indication for liver transplantation in coming decades. It is important that the condition is recognized in children as there is the potential to reverse the process and to avoid the morbidity and mortality associated in later years.
References
1.
Musso G, Gambino R, Cassader M. Recent insights into hepatic lipid metabolism in non-alcoholic fatty liver disease (NAFLD). Prog Lipid Res. 2009;48(1):1–26. PubMed PMID: 18824034. Epub 2008/10/01. eng.PubMed
2.
Moran JR, Ghishan FK, Halter SA, Greene HL. Steatohepatitis in obese children: a cause of chronic liver dysfunction. Am J Gastroenterol. 1983;78(6):374–7. PubMed PMID: 6859017. Epub 1983/06/01. eng.PubMed
3.
Patton HM, Sirlin C, Behling C, Middleton M, Schwimmer JB, Lavine JE. Pediatric nonalcoholic fatty liver disease: a critical appraisal of current data and implications for future research. J Pediatr Gastroenterol Nutr. 2006;43(4):413–27. PubMed PMID: 17033514. Epub 2006/10/13. eng.PubMed
4.
Schwimmer JB, Deutsch R, Kahen T, Lavine JE, Stanley C, Behling C. Prevalence of fatty liver in children and adolescents. Pediatrics. 2006;118(4):1388–93. PubMed PMID: 17015527. Epub 2006/10/04. eng.PubMed
5.
Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, et al. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 2004;40(6):1387–95. PubMed PMID: 15565570. Epub 2004/11/27. eng.PubMed
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


