CHAPTER 85 Nonalcoholic Fatty Liver Disease
In 1980, Ludwig and colleagues from the Mayo Clinic coined the term nonalcoholic steatohepatitis (NASH) to describe a form of liver disease observed in middle-aged patients with abnormal liver biochemical test results and histologic evidence of alcoholic hepatitis but no history of alcohol abuse.1 Much has been learned about NASH since this initial description. NASH is part of the spectrum of nonalcoholic fatty liver disease (NAFLD), which encompasses simple fatty liver, NASH, and NAFLD-associated cirrhosis. NAFLD has emerged as a burgeoning clinical entity, now recognized as an important component of the metabolic syndrome, as well as an exciting area of basic and clinical investigation in the field of hepatology.
NONALCOHOLIC FATTY LIVER AND STEATOHEPATITIS
EPIDEMIOLOGY
The prevalence of NAFLD in the general population is undefined. Several studies have estimated the scope of this disorder in the United States. The Dallas Heart Study of more than 2200 adults documented hepatic triglyceride content by proton magnetic resonance spectroscopy (MRS) and found fatty liver in 31% of participants; the highest prevalence (45%) was among Hispanics.2 Most of the patients with fatty liver by MRS had normal liver biochemical test levels, although the normal range for serum aminotransferase levels in this study was wider than generally accepted. The National Health and Nutrition Examination Survey (NHANES) III, which included more than 15,700 adults, documented unexplained elevations of serum aminotransferase levels, presumably caused by NAFLD, in 2.8% to 5.5% of participants.3,4 Population-based estimates of NAFLD have been reported for other countries as well. These studies have documented NAFLD in 10% to 24% of the population, with the highest prevalence (up to 76%) among obese nondrinkers.5–7 Prevalence estimates vary widely depending on the information available in a given population and the diagnostic criteria that are used to establish the diagnosis (i.e., liver biochemical test levels, radiologic study results, or liver biopsy findings).
Most cases of NAFLD are discovered in the fourth to sixth decades of life, although NAFLD is also described, with increasing frequency, in obese children and adolescents, as well as in older adults. NAFLD may be present long before a diagnosis is established. In early clinical studies, the majority of patients with NAFLD were female; however, subsequent data have suggested that men may be affected as often as women and may be at greater risk for advanced forms of NAFLD, including NASH. The prevalence of NAFLD appears to vary by ethnicity. In the Dallas Heart Study, Hispanics demonstrated the highest prevalence (45%) of NAFLD, compared with 33% for whites and 24% for African Americans. The reasons for racial and ethnic disparities in the prevalence of NAFLD is not known but may be related, at least in part, to racial differences in body fat distribution8 and the prevalence of the metabolic syndrome, which is greatest in people of Hispanic descent.9 Other studies have also shown that African Americans and Mexican Americans have higher frequencies of unexplained serum aminotransferase elevations than do whites.3,4,9,10 Familial clustering of NAFLD may occur,11,12 which likely reflects both genetic and environmental predisposition to the metabolic conditions associated with NAFLD (see later).13
ETIOLOGY
Many agents and conditions have been associated with NAFLD. The causes may be divided into two broad categories (1) drugs and toxins and (2) metabolic abnormalities, either acquired or congenital. Potential causes of NAFLD are listed in Table 85-1.
Table 85-1 Causes of Nonalcoholic Fatty Liver Disease
| Acquired Metabolic Disorders |
| Cytotoxic and Cytostatic Drugs |
| Other Drugs and Toxins |
| Metals |
| Inborn Errors of Metabolism |
| Surgical Procedures |
| Miscellaneous Conditions |
* Tetracycline is cytotoxic by virtue of inhibiting mitochondrial β-oxidation.
Obesity is the condition most often reported in association with NAFLD. Since 1980, the proportion of Americans who are overweight (defined as a body mass index [BMI] > 25 kg/m2) or obese (BMI > 30 kg/m2) has increased markedly. In 2004, 66.2% of Americans adults were classified as overweight or obese, as were 17.4% of children ages 12 to 19 years.14 The health implications of the unremitting obesity epidemic are staggering, and NAFLD is a common byproduct in both adults and children. As noted earlier, most patients with NAFLD are obese. In morbidly obese patients (BMI > 35 kg/m2), including those referred for bariatric surgery, the frequency of NAFLD is as high as 90%, with advanced disease (i.e., NASH) seen in 9% to 40%.15–19 A correlation among BMI, degree of steatosis, and severity of liver injury has been demonstrated in several studies20–22; however, the distribution of body fat may be more important than the total adipose mass for the development of hepatic steatosis. Studies have shown a significant correlation between the risk of the metabolic syndrome, degree of hepatic steatosis, and waist-to-hip ratio, thus highlighting the importance of intra-abdominal or visceral fat as a predictor of NAFLD.23–25
NAFLD also is strongly associated with type 2 diabetes mellitus and glucose intolerance, with or without superimposed obesity.26 Type 2 diabetes mellitus, hyperglycemia, or glucose intolerance has been described in 20% to 75% of adult patients with NASH and may increase the risk of NASH more than twofold compared with that for nondiabetic persons. The presence of NAFLD in diabetic patients may also increase the risk of cardiovascular disease significantly.27 The association between type 2 diabetes mellitus and NAFLD appears strongest in morbidly obese patients.15 NAFLD has been associated with insulin resistance and hyperinsulinemia even in lean subjects with normal glucose tolerance.28 Diabetes mellitus may be an independent predictor of advanced NAFLD, including cirrhosis and hepatocellular carcinoma.29–32
Hyperlipidemia is found in a substantial proportion of patients with NAFLD. Data from the Dallas Heart Study revealed NAFLD in 60% of patients with mixed hyperlipidemia,33 and a study from Korea of potential living liver donors showed that hyperlipidemia was associated with a greater than twofold risk of significant (>30%) steatosis.34 Most patients with NAFLD have multiple risk factors, including central obesity, type 2 diabetes mellitus, and hyperlipidemia, although some affected persons lack all recognized risk factors. NAFLD has been associated with many drugs and toxins and metabolic, surgical, and genetic conditions (see Table 85-1) that have abnormal fat metabolism and mitochondrial injury or dysfunction in common. NAFLD is now recognized as the hepatic component of the metabolic syndrome, which includes hyperlipidemia, glucose intolerance, obesity, and systemic hypertension. The risk and severity of NAFLD increase with the number of components of the metabolic syndrome.28,35
PATHOGENESIS
The pathogenesis of NAFLD is poorly understood, in part because of a lack of suitable animal models that mimic human NAFLD. In light of the variety of conditions that have been associated with NAFLD, it is not surprising that no single pathogenic mechanism has been identified. The prevailing theory is the “two-hit hypothesis,” first proposed by Day and James in 1998.36 This hypothesis states that dysregulation of fatty acid metabolism leads to steatosis, which is the first hepatic insult in NAFLD. Steatosis is associated with several cellular adaptations and altered signaling pathways, which render hepatocytes vulnerable to a “second hit.” The second insult may be one or more environmental or genetic perturbations, which cause hepatocyte necrosis and inflammation and activate the fibrogenic cascade, thereby leading to fibrosis and cirrhosis in a minority of patients with NAFLD.
Hepatic steatosis is the hallmark histologic feature of NAFLD. Normally, free fatty acid (FFA) is supplied to the liver through intestinal absorption (in the form of chylomicron remnants) or from lipolysis of adipose tissue, where FFA is stored as triglycerides. In the liver, FFA is oxidized by mitochondria, esterified into triglycerides, synthesized into phospholipids and cholesteryl esters, and secreted from the liver as very-low-density lipoprotein (VLDL). Under normal circumstances, fatty acid metabolism is under tight regulatory control by catecholamines, glucagon, growth hormone, and insulin. Hepatic triglyceride accumulation occurs when fatty acid metabolism shifts to favor net lipogenesis, rather than lipolysis. This shift occurs when the amount of FFA supplied to the liver from the intestine or adipose tissue exceeds the amount needed for mitochondrial oxidation, phospholipid synthesis, and synthesis of cholesteryl esters. Triglycerides also accumulate in the liver when synthesis of lipoprotein decreases or export of lipids from the liver is impeded (see also Chapter 72).
Current evidence points to insulin resistance and hyperinsulinemia as the primary pathogenic factors in steatosis in most patients with NAFLD. Strong laboratory and clinical evidence supports the association of peripheral insulin resistance and hyperinsulinemia with NAFLD, even in lean patients without obvious glucose intolerance.37–39 The molecular mechanism leading to insulin resistance is complex and not understood completely. In the setting of obesity and hyperinsulinemia, alterations in several molecules, including FFA, tumor necrosis factor-α (TNF-α), membrane glycoprotein PC-1, and leptin, interfere with the insulin signaling pathway. Diabetes mellitus and obesity are associated with increased amounts of FFA in plasma, caused in part by abnormal release of FFA by insulin-resistant adipocytes. Excess FFA contributes to hepatic insulin resistance by down-regulating insulin receptor substrate-1 (IRS-1) signaling.40,41 Insulin resistance and hyperinsulinemia lead to steatosis by means of a number of aberrant mechanisms of FFA disposal. In the liver, insulin stimulates fatty acid synthesis, down-regulates mitochondrial β-oxidation of FFA, blocks the secretion of triglycerides from hepatocytes by increasing intracellular degradation of VLDL and apolipoprotein B-100 (apoB-100), and blocks exocytosis of VLDL-containing vesicles.40,42,43 Also, patients with NASH have impaired hepatic synthesis of apoB-100, which also may contribute to hepatic triglyceride accumulation.44
Insulin resistance in NAFLD may be potentiated by aberrant levels or function of several important peptide mediators secreted by adipocytes, including TNF-α, leptin, and adiponectin. In noninflammatory states, TNF-α is derived from adipose tissue (including adipose tissue macrophages), and plasma levels of TNF-α correlate with body fat mass.45 TNF-α interferes with insulin signaling by down-regulating IRS-1 signaling via serine phosphorylation, likely through activation of stress-related protein kinases including Jun N-terminal kinase (JNK), which plays a key role in obesity-related insulin resistance. Activation of the inhibitor kappa-β kinase (IKK-β)/nuclear factor kappa β (NF-κβ) pathway by FFA may also play a role in reduced hepatic insulin sensitivity,46,47 and may increase production of additional inflammatory cytokines such as TNF-α and interleukin (IL)-6.48 Elevated TNF-α levels have been demonstrated in several studies of NAFLD49–52; however, the independent contribution of TNF-α to the pathogenesis and risk of progression of NAFLD is still unclear.
Adipocytokines are peptides produced by visceral adipose tissue. Adiponectin is secreted by adipocytes in inverse proportion to BMI and is a potent inhibitor of TNF-α. Serum adiponectin levels are reduced in obesity, insulin resistance, diabetes mellitus, and the metabolic syndrome.50 Delivery of recombinant adiponectin to mice fed a high-fat, alcohol-containing diet and to genetically obese (ob/ob) mice dramatically alleviates hepatomegaly, steatosis, inflammation, and elevated liver biochemical test levels in both murine populations.53 These therapeutic effects result in part from the ability of adiponectin to enhance hepatic fatty acid β-oxidation, decrease hepatic triglyceride content, and decrease hepatic insulin resistance. Furthermore, adiponectin suppresses hepatic and plasma concentrations of TNF-α. Studies have reported an inverse relationship between serum adiponectin levels and the degree of steatosis and hepatocyte injury in humans with NAFLD, and this inverse association may be independent of insulin resistance.51,54 Further studies are needed to determine whether an increase in the TNF-α/adiponectin ratio has a primary pathogenic role in the development of steatosis or is more directly correlated with progression from steatosis to steatohepatitis.
Leptin is a satiety hormone, derived from adipocytes, that controls food intake and energy regulation (see Chapter 1). Leptin is intimately involved with insulin signaling and regulation of glucose metabolism in peripheral tissues and may play an important role in regulating the partitioning of fat between mitochondrial β-oxidation and triglyceride synthesis in the liver.55 Severe steatosis and steatohepatitis develop in leptin-deficient (ob/ob) mice. Obesity in humans is associated with relative leptin resistance and high leptin levels, which may contribute to the genesis of steatosis by a negative impact on insulin signaling or may be a consequence of the chronic hyperinsulinemia associated with obesity. Several studies have examined the relationship between serum leptin levels and NAFLD, with conflicting results.56–59 One study has suggested that serum leptin levels in patients with NASH correlate with the severity of hepatic steatosis, independent of BMI, but not with the degree of hepatic inflammation or fibrosis.58 At present, the contribution of leptin to the pathogenesis of NAFLD is unclear.
Although insulin resistance and hyperinsulinemia are clearly pivotal to the development of steatosis, consensus is lacking on the subsequent insults that cause steatosis to progress to steatohepatitis and fibrosis in some patients. Similarities in the histologic features and natural history of alcoholic liver disease and NAFLD suggest that common mechanisms may be involved in the pathogenesis of these disorders. Chronic oxidative stress is believed to be central to the pathogenesis of alcohol-related liver damage. Processes that increase the production of oxidants in the liver during chronic alcohol exposure include the metabolism of ethanol to its reactive intermediate acetaldehyde; induction of microsomal ethanol-oxidizing enzymes, such as cytochrome P450 2E1 (CYP2E1), which generates reactive oxygen species (ROS) that can peroxidize cellular membranes, thereby causing cellular injury60; inhibition of mitochondrial electron transport chain activity; and depletion of mitochondrial glutathione.61 Activation of microsomal enzymes, including CYP2E1, in patients with NAFLD62–63 and mitochondrial production of ROS in murine models of NAFLD64,65 suggest that chronic oxidative stress and lipid peroxidation may also be central to the pathogenesis of NAFLD.
Increased levels of FFA can be directly toxic to hepatocytes through a number of mechanisms. An increased FFA concentration leads to lysosomal destabilization and stimulation of TNF-α.66 FFA also up-regulates cytochrome P450 isoenzymes, leading to enhanced generation of ROS and lipid peroxidation.67 An increased intracellular FFA concentration can lead to sustained up-regulation of peroxisomal proliferator-activated receptor-α (PPAR-α), which promotes fatty acid oxidation and disposal but also may increase oxidative stress through the production of dicarboxylic acid derivatives; PPAR-α also may predispose affected persons to carcinogenesis.44 FFA can be directly toxic to cellular membranes, lead to the formation of toxic fatty acid ethyl ethers, and cause overall disruption of mitochondrial function, thereby overwhelming the overlapping protective mechanisms designed to combat FFA hepatotoxicity.45
Endotoxin and endotoxin-mediated cytokine release are suspected in the pathogenesis of alcoholic steatohepatitis, in which increased serum levels of bacterial endotoxin and lipopolysaccharide (LPS) stimulate hepatic production of TNF-α, IL-6, and IL-8 and activate an inflammatory response that leads to hepatic necrosis (see Chapter 84).61 Bacterial endotoxin also may contribute to the development of NAFLD in some circumstances. Portal endotoxemia was believed to contribute to NASH and hepatic failure associated with surgical jejunoileal bypass (performed in the past to treat obesity), the risk of which was reduced with antibiotics. Yang and colleagues have demonstrated that ob/ob mice with steatosis are highly vulnerable to endotoxin-induced hepatocyte damage, and NASH rapidly develops in these animals after exposure to low doses of bacterial LPS.68 In addition, Zucker diabetic (fa/fa) rats and ob/ob mice demonstrate decreased Kupffer cell function, which may increase the vulnerability of steatotic hepatocytes to TNF-α–mediated liver damage.69 Small studies suggest a possible pathogenic role of bacterial endotoxins in human NAFLD as well,70,71 but these studies are far from conclusive.
A growing body of evidence suggests that mitochondrial changes and altered hepatic energy homeostasis may play roles in the pathogenesis of NAFLD. Studies have shown a decrease in the activity of mitochondrial respiratory chain complexes in steatotic livers, with a concomitant increase in mitochondrial ROS formation; these changes correlate with serum TNF-α levels, insulin resistance, and BMI.72,73 Ob/ob mice have increased levels of uncoupling protein, UCP-2, an inner mitochondrial membrane protein that mediates proton leak, uncouples adenosine triphosphate (ATP) synthesis, regulates ROS production, and may render fatty hepatocytes vulnerable to metabolic stressors65; however, the role of UCP-2 in NAFLD in humans is unknown. Studies have shown that mice and humans with NAFLD have diminished capacity for replenishing ATP stores after ATP depletion. Mitochondrial structural defects may be one cause of reduced ATP stores. Megamitochondria and crystalline mitochondrial inclusions have been identified in patients with NAFLD and may represent an adaptive process to oxidative stress or secondary injury.39,74 Limited data in patients with NASH suggest differential expression of several genes important for proper mitochondrial functioning, including genes involved in ROS scavenging, glucose metabolism, and fatty acid metabolism.75 In addition, mitochondrial DNA damage similar to that found in alcoholic liver disease and Wilson disease also may contribute to the development of NASH. Further animal and human studies are needed to determine whether mitochondrial dysfunction and ATP depletion are causes or consequences of NAFLD.
Fibrosis is a frequent histologic finding in advanced NAFLD but has not been well studied in this disease. Hepatic fibrosis results from activation and proliferation of hepatic stellate cells in the subendothelial space of Disse, with subsequent secretion of extracellular matrix components, including collagen types I and III (see Chapter 90). Factors proposed to initiate and perpetuate the fibrogenic process in stellate cells include inflammatory cytokines, angiotensin, alterations in the extracellular matrix, growth factors, and oxidative stress. In NAFLD, lipid peroxidation products may enhance hepatic production of transforming growth factor-β (TGF-β), which activates stellate cells. Endothelial cells, leukocytes, and Kupffer cells may stimulate the stellate cells to proliferate, possibly through the release of platelet-derived growth factor (PDGF), TGF-β, and other cytokines.76 In addition, hyperinsulinemia and hyperglycemia associated with NAFLD may stimulate release of connective tissue growth factor, an intermediate molecule involved in fibrogenesis.77 Finally, animal data suggest that leptin may perpetuate fibrogenesis in NAFLD by stimulating Kupffer cells and sinusoidal endothelial cells to produce TGF-β.78
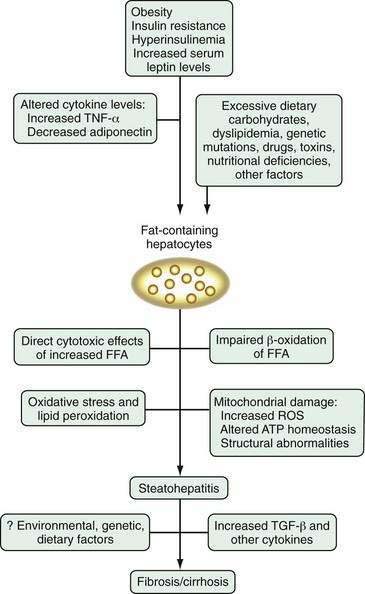
Research into the pathogenesis of NAFLD is proliferating at a rapid pace, but the picture is far from clear (Fig. 85-1). Any one of the putative mechanisms discussed here is unlikely to explain the pathogenesis of NAFLD in all affected patients. More likely, NAFLD develops as a consequence of a “multi-hit” process. The first “hit” is steatosis induced primarily by insulin resistance and hyperinsulinemia. After steatosis develops, a number of factors, including lipid peroxidation, oxidative stress, cytokine alterations, mitochondrial dysfunction, and Kupffer cell activation, may incite an inflammatory response and fibrosis in some patients with genetic or environmental susceptibilities. The exact interplay among these and other factors remains to be elucidated, but understanding of the pathogenesis should be enhanced by the development and refinement of appropriate animal models, including the genetically obese, leptin-deficient ob/ob mouse and Zucker diabetic rats, in which steatosis develops; S-adenosylmethionine (SAM)-deficient mice, in which severe steatohepatitis develops79; the Otsuka-Long-Evans-Tokushima fatty (OLETF) rat model, in which a cholecystokinin-A receptor defect leads to hyperglycemia, obesity, insulin resistance, and hepatic steatosis80; and a high fat-fed rat model in which insulin resistance, elevated serum TNF-α levels, increased oxidative stress, mitochondrial lesions, and early fibrosis develop.81
CLINICAL, LABORATORY, AND IMAGING FEATURES
The clinical and laboratory features of NAFLD are summarized in Table 85-2. NAFLD usually is discovered incidentally because of elevated liver biochemical test levels or hepatomegaly noted during an evaluation for an unrelated medical condition. Most patients with NAFLD are asymptomatic, but some may describe vague right upper quadrant pain, fatigue, and malaise. Hepatomegaly has been described in up to 75% of patients with NAFLD but often is difficult to appreciate on physical examination because of obesity. Stigmata of chronic liver disease, such as splenomegaly, spider angiomata, and ascites, are rare, except in patients with NAFLD-associated cirrhosis.
Table 85-2 Clinical and Laboratory Features of Nonalcoholic Fatty Liver Disease
| SYMPTOMS | SIGNS | LABORATORY FEATURES |
|---|---|---|
| Common | ||
| None (48%-100% of patients) | Hepatomegaly | |
| Uncommon | ||
ANA, antinuclear antibodies; ALT, alanine aminotransferase; AST, aspartate aminotransferase.
Elevated liver biochemical test levels may be found in up to 50% of patients with simple steatosis and are present in approximately 80% of patients with advanced NAFLD. A mild-to-moderate (1.5- to 4-fold) elevation of the serum aspartate aminotransferase (AST) or alanine aminotransferase (ALT) level, or both, is usual, and levels seldom exceed 10 times the upper limit of normal. The serum ALT level usually is greater than the AST level, in contrast with the pattern of alcoholic hepatitis, in which the AST level is at least twofold higher than the ALT level (see Chapters 73 and 84). The alkaline phosphatase and gamma glutamyl transpeptidase (GGTP) levels may be elevated, but the serum bilirubin level, prothrombin time, and serum albumin level typically are normal, except in patients with NAFLD-associated cirrhosis. Up to one fourth of patients with NAFLD may have antinuclear antibodies (ANA) in low titers (less than 1 : 320).82 Antimitochondrial antibodies (AMA) and hepatitis B surface antigen are not detected. Antibody to hepatitis C virus (anti-HCV) must be absent to implicate NAFLD as the sole cause of abnormal liver biochemical test levels; however, steatosis, often in association with visceral obesity, frequently accompanies HCV infection and may be associated with a more aggressive course (see Chapter 79).83 Serum ceruloplasmin and α1-antitrypsin levels are within normal limits. Serum and hepatic iron levels may be elevated in patients with NAFLD. In particular, the serum ferritin level may be elevated in 20% to 50% of patients with NAFLD and may be a marker of more advanced disease.29,84
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


