Motility Disorders
Introductory Comments
Normal gastrointestinal (GI) motility depends on intact neuromuscular functions that consist of both intrinsic and extrinsic innervation. Extrinsic control of peristalsis includes the sympathetic (thoracolumbar) and parasympathetic (vagal) innervation in the ganglionated plexuses. Intrinsic control includes the enteric nervous system (ENS), smooth muscle cells, and interstitial cells of Cajal (ICCs), with the latter serving as both pacemaker cells and as intermediaries of enteric innervation (1,2). Motility disturbances constitute a complex array of clinical and pathologic disorders that result from neural, muscular, neuromuscular, or ICC abnormalities (Tables 10.1, 10.2, 10.3, and 10.4). Intestinal neuropathies appear to be more common than intestinal myopathies. Motility disorders occur at any age and may be primary or may complicate systemic diseases. Primary motility diseases more typically affect children than adults. Conversely, secondary conditions, such as scleroderma-associated myopathy, diabetic neuropathy, drug-induced damage, or viral infections, more frequently affect adults. Primary motility disorders may be familial or sporadic. They may remain limited to the gut, as in Hirschsprung disease (HD), or they may be part of a generalized peripheral autonomic neuropathy, as in familial visceral neuropathy. Familial disorders are inherited as both autosomal recessive and autosomal dominant diseases.
The clinical and/or pathologic findings of gastrointestinal motility disorders may be subtle or dramatic. They can present as dysphagia, nausea, vomiting, diffuse esophageal spasm, gastroparesis, intestinal pseudo-obstruction, constipation, or intestinal diverticulosis. Intestinal pseudoobstruction is defined as a rare, severe disabling disorder characterized by repetitive episodes or continuous symptoms and signs of bowel obstruction, including radiographic documentation of a dilated bowel with air–fluid levels in the absence of a fixed, lumen-occluding lesion (3). In contrast, Ogilvie syndrome is a term used synonymously with acute colonic pseudo-obstruction. The terms megaesophagus, megaduodenum, megajejunum, megacolon, and megarectum describe visceral enlargement of each of these anatomic sites. There is no agreement on the criteria for the minimum diameters of the dilated gastrointestinal segments.
Small intestinal pseudo-obstruction leads to diarrhea, malabsorption, and steatorrhea secondary to bacterial overgrowth. Some patients become malnourished with extreme weight loss.
Extraintestinal manifestations depend on the nature of the underlying disease; some help define specific syndromes. Features suggesting autonomic dysfunction include postural dizziness, difficulties in visual accommodation to bright light, and sweating abnormalities. Recurrent urinary infections and difficulty emptying the urinary bladder suggest a general visceral neuromyopathic disorder. Patients should also be questioned about drug use. Bedridden patients, such as those with dementia, stroke, and spinal cord injuries, are particularly prone to developing megacolon and a chronic pseudo-obstruction.
Motility disorders are clinically diagnosed using specific physiologic measurements of gastrointestinal motor function, including scintigraphy, gastroduodenojejunal manometry, and surface electrogastrography. The clinician will also often seek the pathologist’s assistance to rule out the presence of infiltrative lesions, such as amyloidosis or connective tissue diseases, or to document the presence of neuromuscular abnormalities.
Even though the clinical or gross findings may be dramatic, the histologic features are often inconspicuous, and they may also overlap with nonspecific neural and/or muscular histologic abnormalities accompanying other conditions, such as carcinoma or previous surgery. Additionally, some patients with clinically evident motility disorders may have histologic abnormalities that have not been well described or placed into specific syndromes. In other patients, there may be neurotransmitter alteration that may or may not associate with morphologic abnormalities. These most commonly involve alterations in the nitrergic neural system or changes in vasoactive intestinal polypeptide (VIP) or substance P (SP)-containing nerves. Changes may also be present in the muscle cells or ICCs.
Histologic examination using conventional hematoxylin and eosin (H&E) stains is usually augmented with the use of special stains or ultrastructural examination (Table 10.5). Some histologic changes are so subtle that they require precise neuronal counting to document their presence. However, this is fraught with problems, since the number of
nerves and ganglia vary with age, location, other disease processes, and section thickness. A method for rapidly assessing enteric ICCs and neuronal morphology has been developed using antibodies to c-kit and NF 68. The procedure takes approximately an hour to perform (4). It remains to be seen whether it will be widely adopted.
nerves and ganglia vary with age, location, other disease processes, and section thickness. A method for rapidly assessing enteric ICCs and neuronal morphology has been developed using antibodies to c-kit and NF 68. The procedure takes approximately an hour to perform (4). It remains to be seen whether it will be widely adopted.
TABLE 10.1 Gastrointestinal Neural Disorders | |
|---|---|
|
TABLE 10.2 Gastrointestinal Muscular Disorders | |
|---|---|
|
Treatment of motility disorders ranges from dietary changes or pharmacologic treatment to surgery or intestinal transplantation. Prokinetic drugs such as cisapride, metoclopramide, and octreotide benefit some patients. Patients with acute colonic pseudo-obstruction may benefit from neostigmine treatment (5). Bowel decompression through gastrostomy and jejunostomy may help some patients. Small bowel transplantation is the only definitive cure for patients with chronic pseudo-obstruction. Candidates for transplantation include those receiving total parenteral nutrition with frequent episodes of sepsis, limited intravenous access to nutritional support, or impending liver failure. However, small bowel transplantation tends to be challenging in this clinical setting.
Normal Neuromuscular Structure
Muscles
The muscularis propria is a continuous structure composed of two smooth muscle layers that extend from the upper esophagus to the anal canal (Fig. 10.1). The only exception to this occurs in the stomach, where three muscle layers are
present. At the junctions between adjacent organs, the muscular coat rearranges to form sphincters including the pharyngoesophageal, esophagogastric, pyloric, ileocecal, and anal sphincters. The function of these sphincters is based on physiologic and pharmacologic characteristics of the musculature and on their innervation. The muscle fibers are usually arranged in a concentric circular fashion in the inner muscular layer (the circular layer), whereas the outer muscle fibers are arranged longitudinally (the longitudinal layer). In the cecum and in parts of the colon, the longitudinal muscle is extremely attenuated except in the regions where it forms thick cords (i.e., the taeniae coli).
present. At the junctions between adjacent organs, the muscular coat rearranges to form sphincters including the pharyngoesophageal, esophagogastric, pyloric, ileocecal, and anal sphincters. The function of these sphincters is based on physiologic and pharmacologic characteristics of the musculature and on their innervation. The muscle fibers are usually arranged in a concentric circular fashion in the inner muscular layer (the circular layer), whereas the outer muscle fibers are arranged longitudinally (the longitudinal layer). In the cecum and in parts of the colon, the longitudinal muscle is extremely attenuated except in the regions where it forms thick cords (i.e., the taeniae coli).
TABLE 10.3 Disorders Involving the Interstitial Cells of Cajal | |
|---|---|
|
TABLE 10.4 Secondary Neuromuscular Disorders | |
|---|---|
|
Circular muscle, from the esophagus to the internal anal sphincter, behaves as an electrical syncytium resulting from nexuses between the plasma membranes of contiguous muscle fibers. These nexuses function as intracellular pathways for excitation conduction between adjacent cells. Even in the absence of neural influences, these syncytial properties allow three-dimensional spread of excitation (6). Smooth muscle cells also contain gap junctions or nexuses that electrically couple adjacent cells (6). The musculature also contains the ICCs, which act as pacemakers for the GI muscle (see below) and facilitate active propagation of electrical events.
TABLE 10.5 Markers Useful in Evaluating Intestinal Motility Disorders | ||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
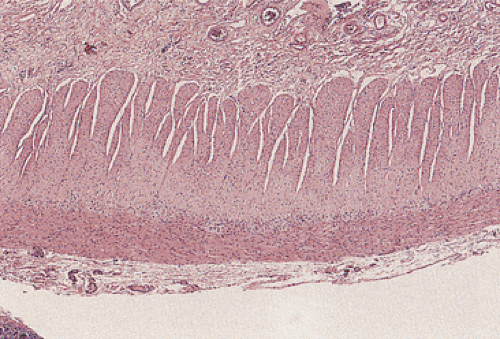 FIG. 10.1. Hematoxylin and eosin–stained section demonstrating the circular and longitudinal smooth muscle layers of the muscularis propria. This layer is present throughout the gastrointestinal tract. |
The cells of the muscularis contain numerous receptors, allowing them to respond to neural signals, as well as other stimulatory and inhibitory signals during the digestive process. Contraction of the circular layer constricts the lumen; contraction of the longitudinal layer shortens the digestive tube. When the bowel becomes obstructed or the intestinal lumen distends on a persistent basis, the muscle increases in volume through both hypertrophy and hyperplasia. Smooth muscle hyperplasia also follows myenteric ablation. Obstruction results in a number of changes to both the muscular and neural layers.
Innervation
The ENS is the most complex portion of the peripheral nervous system. Three divisions of the nervous system (sympathetic, parasympathetic, and enteric) contribute to the neural control of at least four physiologic effector systems: The visceral smooth muscle responsible for motility and sphincteric functions, the mucosa responsible for gastric acid secretion and intestinal fluid and electrolyte homeostasis, the immune cells responsible for mucosal immunity, and the vasculature. Complex reflex activities involving GI motility, ion transport, and mucosal blood flow all occur in the absence of extrinsic autonomic and sensory input.
Functionally, neurons of the ENS fall into five types: (a) motor neurons, efferent or effector neurons acting to control smooth muscle tone in the wall of the gut; (b) vasomotor neurons, which control vascular muscle tone; (c) secretory neurons, other effector neurons regulating exocrine and endocrine secretion; (d) sensory neurons, which carry sensory information to the central nervous system; and (e) interneurons, which provide communication between neurons and the gut wall (Fig. 10.2). These intermingle in the myenteric and submucosal ganglia.
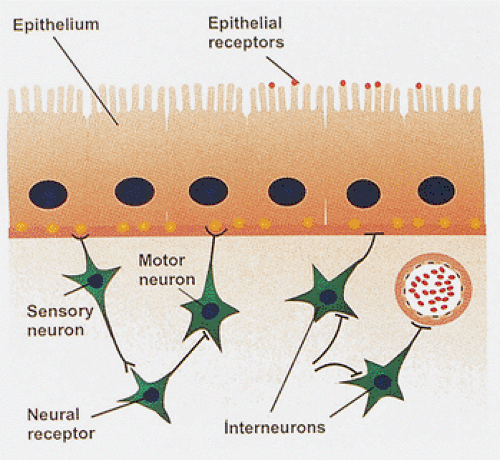 FIG. 10.2. Diagram showing the interactions of sensory neurons and motor neurons with interneurons, epithelial cells, blood vessels, and portions of the muscularis mucosae. |
The ENS consists of three ganglionated plexuses in the gut wall: The myenteric (Auerbach) plexus located between the longitudinal and circular muscle layers of the muscularis propria and the submucosal (Meissner) plexus found in the submucosa. A third plexus, which derives from extrinsic nerves, occurs in the inner quarter of the circular muscle coat adjacent to the submucosa and is rich in ICCs (7). These plexuses extend uninterrupted from the esophagus to the anus, innervating the mucosa, muscle layers, and blood vessels. Interconnections between submucosal and myenteric plexuses coordinate motility and ion transport. Intramural ganglia contain nerve cells, glia (Fig. 10.3), and a neuropil formed by neuronal processes (some of extrinsic origin and others issued by intrinsic neurons) and glial processes. Since neural fibers from many different origins exist within the neuropil, the neuronal organization is extremely complex with controls coming from both the intramural and extramural ganglia.
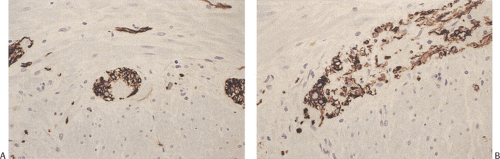 FIG. 10.3. Myenteric plexus stained with an antibody to glial fibrillary acidic protein showing the large glial component. A: The plexus in cross section. The central unstained area corresponds to a ganglion cell. B: Longitudinal section of plexus. The unstained portions correspond to neurons. |
Generally, the longitudinal musculature is poorly innervated (6). The innervation of the musculature is particularly dense at the level of the sphincters. The anal sphincter has the densest adrenergic innervation found in the GI tract. Most of its fibers originate from the superior mesenteric ganglion, but some derive from ganglionic neurons of the sacral sympathetic chain. Adrenergic fibers are also plentiful in the sphincter of Oddi. Nerve fibers containing VIP are numerous in the musculature of the gastroesophageal junction, the pylorus, and the sphincter of Oddi.
It is estimated that there are more neurons in the GI tract than there are in the spinal cord (8). Neurons of the ENS may be classified by their transmitters. Utilizing this classification, there are at least five types of neurons: (a) cholinergic (acetylcholine containing); (b) adrenergic (norepinephrine containing); (c) GABAergic (γ-aminobutyric acid containing); (d) peptidergic (peptide containing); and (e) nitrergic. The intrinsic nerve fibers are nonadrenergic noncholinergic (NANC) peptidergic nerves. The myenteric plexus neurons may also be viewed as argyrophilic and argyophobic cells based on their silver staining characteristics. Argyrophilic cells are often multiaxonal. The argyophobic cells are cholinergic nerve fibers that may directly contact the muscle cells; argyrophilic neurons do not commonly contact muscle cells. Myenteric nerve trunks consist of both extrinsic (sympathetic and parasympathetic) and intrinsic nerve fibers. In most GI regions, the ENS independently regulates many gut functions including motility, vascular tone, secretion, and release of hormones, although the central nervous system modulates the reflexes (9,10).
Normal bolus propagation depends on both cephalic excitation of gut segments producing propulsive pressure and on caudal relaxation and reduction in flow resistance.
Further, active relaxation of the sphincters is critical to prevent functional obstruction of these regions. Motor neurons of the myenteric plexus, which can be excitatory or inhibitory in nature, are responsible for the immediate neural control of gut tone. The major excitatory motor pathway involves acetylcholine, enkephalin, and tachykinins such as substance P and neurokinin A, whereas the main inhibitory transmitters are nitric oxide (NO), VIP, and adenosine triphosphate. Neural NO is produced by neural nitric oxide synthase (nNOS) (11). VIP and NO are cotransmitters in the NANC nerve–mediated smooth muscle relaxation, and part of VIP actions may be mediated by NO (Fig. 10.4) (12). The nitrergic neurons lie within the myenteric plexus. NO is also produced by the smooth muscle cells (13). The pyloric sphincter has the highest nNOS levels (13). The internal anal sphincter also contains high levels of the enzyme (13).
Further, active relaxation of the sphincters is critical to prevent functional obstruction of these regions. Motor neurons of the myenteric plexus, which can be excitatory or inhibitory in nature, are responsible for the immediate neural control of gut tone. The major excitatory motor pathway involves acetylcholine, enkephalin, and tachykinins such as substance P and neurokinin A, whereas the main inhibitory transmitters are nitric oxide (NO), VIP, and adenosine triphosphate. Neural NO is produced by neural nitric oxide synthase (nNOS) (11). VIP and NO are cotransmitters in the NANC nerve–mediated smooth muscle relaxation, and part of VIP actions may be mediated by NO (Fig. 10.4) (12). The nitrergic neurons lie within the myenteric plexus. NO is also produced by the smooth muscle cells (13). The pyloric sphincter has the highest nNOS levels (13). The internal anal sphincter also contains high levels of the enzyme (13).
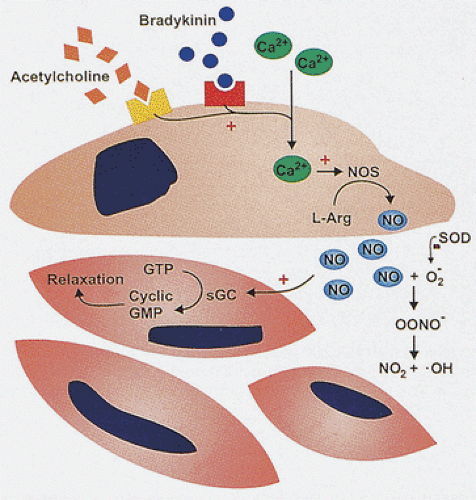 FIG. 10.4. Nitric oxide (NO) mediates vascular relaxation. NO is produced in endothelial cells by the action of calcium-dependent nitric oxide synthase (NOS). Mediators such as acetylcholine and bradykinin stimulate its production. NO acts on smooth muscle cells through a process involving conversion of guanosine triphosphate (GTP) to cyclic guanosine monophosphate (GMP), affecting relaxation. NO released into the interstitium may produce damaging free radicals after interacting with superoxide molecules. |
Interstitial Cells of Cajal
ICCs act as the pacemaker cells controlling smooth muscle contractions (14). They also act as spatial coordinators (15) and intermediaries in the neural control of gut muscular activity (16). ICCs are present in the esophagus, stomach, small and large intestine, and anorectum (17,18). There are distinct subpopulations of ICCs including intramuscular ICCs, myenteric plexus ICCs, and submucosal ICCs (19). In the esophagus, gastric cardia, and fundus, they are present in the muscularis propria but not in the myenteric plexus or submucosa. In the gastric nonfundic corpus and pylorus and in the intestines, they are present in the myenteric plexus. Submucosal ICCs are seen in the small and large intestines. ICCs express the proto-oncogene c-kit (1), making them easy to visualize with immunohistochemical stains. ICCs have long cell processes and show bipolar or multipolar configurations.
Development of the Enteric Nervous System
The ENS derives from vagal and sacral regions of the neural crest (10). Crest-derived cells migrate to the gut at a stage when they are morphologically indistinguishable from surrounding mesenchymal cells. Interactions of the neural crest cells with components of the extracellular matrix play a role in specifying where crest-derived cells migrate. Precursors arriving in the gut are multipotential and the enteric microenvironment to which they migrate dictates the phenotypic differentiation of developing enteric ganglion cells.
Enteric neuronal migration and differentiation involves complex interactions of lineage-determined microenvironmental elements, including transcription factors, tyrosine kinase receptor oncogenes and their ligands, the extracellular matrix, and specific adhesion molecules (10,20,21,22). Among some of the more important ligands of the tyrosine kinase receptor ligands are the neurotrophin family of growth factors that promote the differentiation, growth, and survival of various neurons (23). Glial cell line–derived neurotrophic factor (GDNF) and neurturin play an important role in the migration of enteric neuron precursors into and along the small and large intestines and the esophagus, a process that is RET dependent (24,25). GDNF, which acts as a chemoattractant for enteric neural cells, promotes migration of neural crest cells throughout the gastrointestinal tract, attracting them to this site and preventing them from straying out of the gut (26). Analysis of animal models (transgenic and knockout mice) has played an important role in identifying the genes critical for ENS development (21,27,28,29) (Table 10.6).
Vagal nerve trunks can be found in the upper esophagus by the fifth fetal week. Nerve trunks extend on the outer surface of the gut wall and neuroblasts are found along the gastric cardia by the sixth week, the cephalic limb of the midgut by the seventh week, and the entire gut up to the distal half of the colon and rectum by the eighth week. By the 12th week, they have migrated as far as the rectum. The myenteric plexus forms just outside the circular muscle coat, which then develops its longitudinal muscle coat (30). Neuroblasts migrate from the myenteric plexus to form the submucosal plexus; this is completed during the third and fourth months (30). Following neural crest cell migration, the cells proliferate and the neurons
mature (Fig. 10.5), developing processes and synapses that allow for communication within ganglia, between ganglia, and between ganglia and smooth muscle cells. Interruption of this process at different stages produces various myenteric plexus abnormalities ranging from agenesis to incomplete maturation. Interruption of the orderly cranial–caudal migration of nerves and neuroblasts explains the variable length of aganglionic segments in Hirschsprung disease.
mature (Fig. 10.5), developing processes and synapses that allow for communication within ganglia, between ganglia, and between ganglia and smooth muscle cells. Interruption of this process at different stages produces various myenteric plexus abnormalities ranging from agenesis to incomplete maturation. Interruption of the orderly cranial–caudal migration of nerves and neuroblasts explains the variable length of aganglionic segments in Hirschsprung disease.
TABLE 10.6 Genes Involved in Enteric Nervous System (ENS) Development | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
At birth, normal enteric ganglia contain both mature and immature neurons. Premature infants have more immature neurons than term infants. Mature neurons are larger than immature neurons and they have a distinct cell membrane, a vesicular nucleus, and a large amount of basophilic cytoplasm. Immature neurons are small cells with dark nuclei, clumped chromatin, and scant cytoplasm (Fig. 10.6). Neural stains highlight immature neurons. The normal mature colon contains 7 ganglion cells/mm of myenteric plexus, 3.6/mm in the jejunum, and 4.3/mm in the ileum in 3 micron sections (31). Ganglion cells lie approximately 1 mm apart; they may occur in clusters of from one to five cells in normal adults (32). Normal neonates often have plentiful, prominent ganglion cells, but they appear small when they are immature (30).
Most patients with congenital myenteric plexus abnormalities fall into one of five categories: (a) aganglionosis, (b) hypoganglionosis, (c) hyperganglionosis, (d) ganglionic immaturity, and (e) poorly classified abnormalities. The pathogenesis of developmental disorders of the ENS results from genetic defects, failed neural crest migration or differentiation, anoxia, or inflammation. Developmental neural diseases occur alone or coexist with systemic disorders such as neurofibromatosis.
Hirschsprung Disease
Synonyms for Hirschsprung disease include aganglionic megacolon, congenital megacolon, and aganglionosis.
HD is a congenital disorder characterized by intestinal megacolon, neural hyperplasia, and aganglionosis. Several forms of the disease are recognized.
Classic form: The aganglionic segment begins in the distal colorectum and extends proximally for variable distances into the adjoining proximally dilated bowel (Fig. 10.7).
Short segment form: The aganglionic segment involves several centimeters of the rectum and rectosigmoid. The aganglionic segment may be as short as 3 cm, so that this variant may be missed if the biopsy is taken too high above the pectinate line. This form affects 67% to 90% of patients.
Long segment form: The aganglionic segment extends beyond the sigmoid and involves a variable length of colon but does not extend beyond the cecum. This form affects<10% of patients (33).
Total colonic aganglionosis: The entire colon is involved along with variable lengths of ileum, jejunum, and even the stomach. This form affects 2.6% to 14.9% of cases. This form almost always presents in the first weeks of life.
Zonal colonic aganglionosis (synonym: Skip segment HD): A short bowel segment is involved in this form of HD (Fig. 10.8). Ganglion cells are present, both proximal and distal to the aganglionic segment. (This form of the disease may in fact not be HD, but the result of intrauterine injury.)
HD affects 1 in every 5,000 to 30,000 live births; 80% of patients are male (34). Approximately 4% to 6% of cases
are familial (34), especially when the megacolon extends to the cecum. Five percent of patients have an affected sibling (35).
are familial (34), especially when the megacolon extends to the cecum. Five percent of patients have an affected sibling (35).
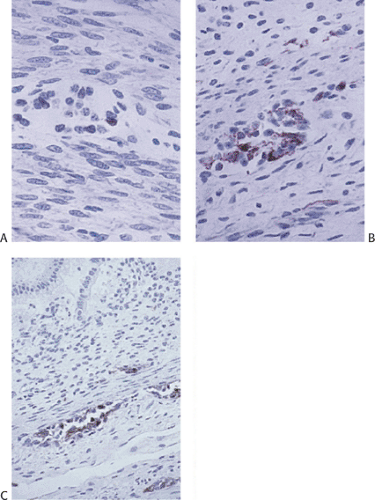 FIG. 10.5. Fetal plexus stained with an antibody to neurofilament protein. A: A 13-week fetus with only rare cells immunoreactive for neurofilament protein. Note the lack of dendritic processes. B: A 15-week fetus showing further development of the neuroblastic cells and the beginning of the appearance of dendritic processes. C: A 17-week fetus with better developed processes and the beginning appearance of submucosal plexuses with fibers having neuronal extensions. |
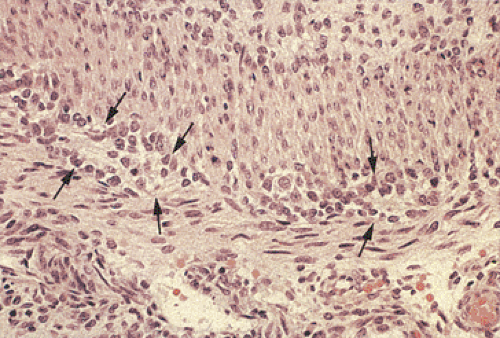 FIG. 10.6. Fetal gut stained with hematoxylin and eosin. Note the presence of immature neuroblastlike cells without clearly identifiable ganglia (arrows). |
Etiology and Pathophysiology of the Disease
HD is a heterogeneous genetic disorder with autosomal dominant, autosomal recessive, and polygenic forms of inheritance. Current etiologic hypotheses revolve around two major schools of thought: Arrested neuroblast migration and intestinal microenvironmental abnormalities that cause failed neuronal differentiation. Specific genetic mutations are present in about 50% of cases. Much of the phenotypic variability of the disease relates to the biologic complexity underlying the normal development of the ENS and the diversity of the molecular alterations that have been identified. Thus, no single genetic abnormality accounts for the development of the disease.
There are several susceptibility genes for HD (Table 10.7) (36,37,38,39,40,41,42,43,44). Other genes that may be abnormal include endothelin-converting enzyme and the transcription factor Sox 10 (40,41). Several genes may modify the severity of the HD phenotype in patients with or without coexisting intestinal neuronal dysplasia. These may lie near 21q22 (43) and may account for the prevalence of HD among patients with trisomy 21 (43). The nerves in HD also fail to express the trkC tyrosine receptor and its ligand neurotrophin, suggesting
that neurotrophic factors that are critical for cellular survival and differentiation may play a role in the pathogenesis of HD (45).
that neurotrophic factors that are critical for cellular survival and differentiation may play a role in the pathogenesis of HD (45).
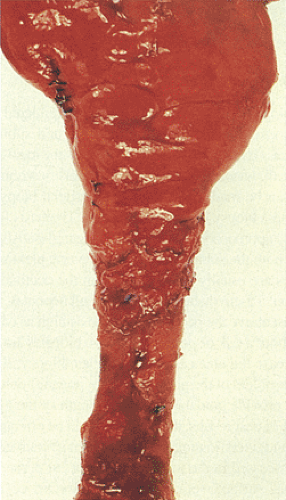 FIG. 10.7. Gross specimen of a patient with Hirschsprung disease. Note the contracted distal bowel, which tapers distally. The proximal bowel is dilated. |
The ret protein is a tyrosine kinase receptor with extracellular cadherinlike and cysteine-rich domains, a transmembrane domain, and an intracellular tyrosine kinase domain (46). Point mutations in the RET gene give rise to HD, multiple endocrine neoplasia (MEN) types IIA and IIB, and familial medullary thyroid carcinoma (47). In the case of MEN-II, the RET mutations are activating, enhancing the function of the encoded protein, whereas in HD, the mutations are inactivating, leading to loss of its function (48). A ret codon 618 ser mutation could predispose patients with MEN-IIA to HD (49).
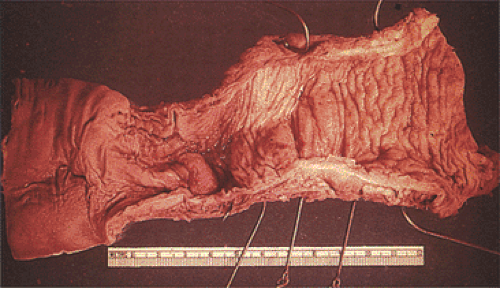 FIG. 10.8. Zonal aganglionosis. The distal bowel lies to the right of the photograph and the proximal bowel to the left. The aganglionic segment is the contracted segment in the middle. The proximal bowel appears dilated and featureless, with loss of the mucosal markings. |
More than 50 RET mutations, including missense, nonsense, deletion, and insertion mutations, have been described in HD. These mutations occur throughout the gene, without any mutational hot spots (50). They fall loosely into two groups: Frame shift or missense mutations that disrupt the structure of the intracellular tyrosine kinase domains and missense mutations in exons 2, 3, 5, or 6 of the extracellular domain (51). Patients with mutations of the intracellular domain have either short segment or long segment HD, whereas those with mutations in the extracellular domain all have long segment HD. RET mutations are more
common among familial cases (50%) than among sporadic cases (15% to 33%) (50). Immunohistochemically, the bowels of patients with HD show reduced ret protein expression.
common among familial cases (50%) than among sporadic cases (15% to 33%) (50). Immunohistochemically, the bowels of patients with HD show reduced ret protein expression.
TABLE 10.7 Mutations Present in Hirschsprung Disease (HD) | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
In addition to the presence of specific RET mutations that may lead to the development of HD, there are also RET intragenic polymorphisms that lead to various clinical phenotypes (52). The c135G/A polymorphism or sequence variations in linkage disequilibrium with this polymorphism modulate the phenotypic influences of RET germline mutations. The c135A variant associates more commonly with short segment disease when it is on the same chromosome with the germline mutation (52).
Alterations in the intrinsic gastrointestinal innervation contribute to the clinical and pathologic features of HD. VIP and NO, components of the NANC system that relax smooth muscle and form part of the inhibitory component of the peristaltic reflex, are absent (53,54,55). However, extrinsic parasympathetic, cholinergic, and sympathetic adrenergic innervation persist. As a result, the distal aganglionic bowel is under constant, unopposed extramural stimulation so that it becomes narrowed, spastic, and unable to support peristalsis. There are also conflicting reports on the ICCs in HD. Some authors find normal numbers of ICCs, whereas others suggest that they are decreased in number.
The pathogenesis of the enterocolitis, which affects some patients, is not well understood. It is likely to result from the toxemia due to bacterial stasis in the dilated colonic lumen. Risk factors for enterocolitis include a delayed diagnosis of HD, long segment disease, family history for HD, female gender, and trisomy 21 (56).
Clinical Features
HD is the most common form of congenital intestinal obstruction, often presenting within the first 24 to 48 hours of life. Up to 80% of cases are diagnosed during the first year of life; 10% first present in adults. Typically, the lack of propulsive movements and inhibitory reflexes in an intestinal segment leads to abdominal distension, vomiting, severe constipation, and marked dilation of the proximal ganglionic segment. Infants with obstruction but without megacolon should be suspected of having HD involving the entire colon. Reduced food intake and malabsorption result in failure to thrive. As the nutritional status deteriorates, infections may worsen the underlying motility problem. Some patients develop mucosal prolapse at the junction of the ganglionic and aganglionic bowel due to differential luminal pressures in these bowel segments. Mucosal prolapse is more prominent in older patients and correlates with disease duration. HD patients may also present in the neonatal period with perforation due to a coexisting necrotizing enterocolitis. The enterocolitis has an ischemic basis and is characterized by mucosal ulceration, colonic bleeding, perforation, sepsis, and toxemia. Patients with trisomy 21 exhibit an increased incidence of HD-associated enterocolitis (56).
Ten to fifteen percent of patients have associated congenital anomalies or other diseases (Table 10.8). Ten percent of patients have Down syndrome; 5% have other serious neurologic abnormalities (57).
Pathologic Findings
The widely dilated, fluid-filled, hypertrophic colon empties into a funnel-shaped transitional zone extending to the anus (Fig. 10.7). Plain abdominal films may show air–fluid levels. The anal canal and rectum are small and empty, and the anal sphincter is tight. In adults, an abrupt, smooth rectal transition zone with proximal colonic dilation, in the setting of an appropriate clinical history, suggests the diagnosis.
A diagnosis of HD is usually made on a suction rectal biopsy containing both the mucosa and submucosa since the aganglionosis coincides closely in both the submucosal and myenteric plexuses. Biopsies are usually taken 2 cm from the pectinate line and at about a 5-cm distance. In very small neonates, the biopsy is taken just above the pectinate line and as high as can be taken safely without risking perforation. Two biopsies are preferred to increase the chances of an adequate biopsy, to overcome the possibility of a hypoganglionic segment, and to provide guidance as to the length of the aganglionic segment. Full-thickness rectal biopsy is reserved for patients in whom the diagnosis cannot be made with a more superficial biopsy.
TABLE 10.8 Other Abnormalities Affecting Patients with Hirschsprung Disease | |
|---|---|
|
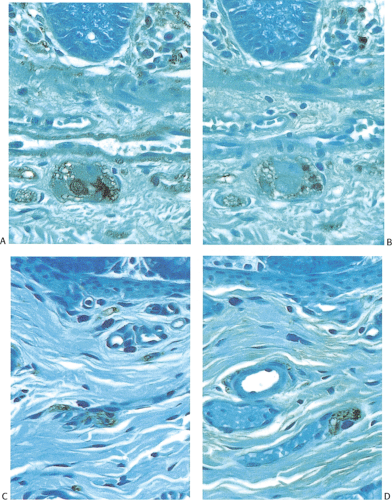 FIG. 10.9. A: Section of normal colon stained with anti–neuron-specific enolase (NSE) to highlight the submucosal ganglion. B: Serial section stained with anti-S100. The ganglion cells are negative. C: Hirschsprung disease stained with anti-NSE. Ganglion cells are absent, and there is nerve fiber proliferation. This is seen better at higher power magnification. D: Serial section stained with anti-S100. Nerve fibers are positive. |
Typical features of HD include aganglionosis (Fig. 10.9) and increased numbers of hypertrophic, nonmyelinated, submucosal, and myenteric plexus cholinergic nerves (part of the extrinsic parasympathetic innervation) (Fig. 10.10). While neural hyperplasia is characteristic of HD, it should be noted that neural hyperplasia is present in other disorders (Table 10.9). Ganglion cells are absent from both plexuses in the distal narrowed bowel and are decreased in number in the first few centimeters of the transitional zone. An increase in the number of ganglion cells occurs as one progresses proximally into the funnel-shaped transitional zone and into the normally innervated bowel. The transitional zone usually occurs over a short distance with ganglia appearing almost simultaneously in both the myenteric and submucosal
plexuses. Some patients have longer transitional zones than others; prominent nerve trunks may be present for several centimeters. The transitional zone may contain abnormally shaped ganglia. Some transition zones show features of colonic neuronal dysplasia (see below).
plexuses. Some patients have longer transitional zones than others; prominent nerve trunks may be present for several centimeters. The transitional zone may contain abnormally shaped ganglia. Some transition zones show features of colonic neuronal dysplasia (see below).
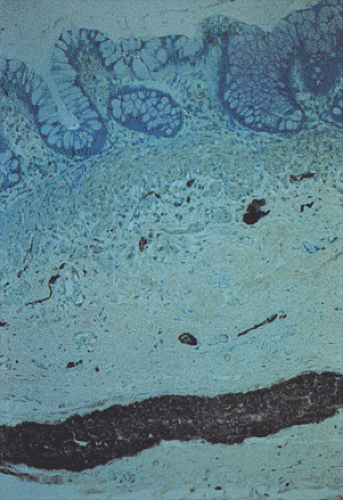 FIG. 10.10. Hypertrophic nerve in Hirschsprung disease stained with an antibody to S100. |
In premature infants it may be difficult to recognize the immature ganglion cells due to their small size and inconspicuous nuclei (Fig. 10.6). They form rosettelike structures arranged around a central neuropil-type matrix producing a horseshoelike structure. The immature ganglia may mimic macrophages, smooth muscle cells, and Schwann cells. The ganglia may be highlighted with special stains. However, it should be kept in mind that immature ganglia may fail to stain with ganglionic markers. These patients also have decreased numbers of synaptophysin-positive synapses in the circular and longitudinal muscle layers in the transitional segments and in aganglionic segments (58) and the adrenergic and peptidergic innervation of aganglionic gut is abnormal. Patients also have a relative loss of ICCs (59), although this finding is not present in all studies (60). Increased numbers of mast cells, often in direct contact with the hypertrophic nerves, are present. These produce nerve growth factor, which stimulates neural growth.
TABLE 10.9 Conditions Associated with Hyperplasia of the Myenteric Plexus | |
|---|---|
|
Frequently, the bowel wall proximal to the aganglionic segment is biopsied at the time of surgery to ensure that the proximal resection margin is normal. Submucosal nerve trunks >40 μm in diameter or abnormal-appearing ganglia strongly correlate with abnormal innervation and aganglionosis (Fig. 10.11) (61). If hypertrophic nerve trunks or abnormal ganglion cells are present in frozen sections, the surgeon should extend the resection proximally and monitor it with additional frozen sections to identify a region that contains completely normal neural structures in order to prevent recurrent disease. Once resection specimens are received, the extent of the aganglionosis should be determined and the status of the proximal margin should be ascertained, if this was not done intraoperatively. Patients who develop postoperative symptoms may have either a retained portion of the transitional zone with neuronal dysplasia or an aganglionic segment or they may have developed an acquired disorder secondary to postoperative ischemia or infection.
If enterocolitis develops, the histology may include crypt dilation with mucin depletion, cryptitis, crypt abscesses, mucosal ulcers, transmural necrosis, and perforation. Enterocolitis affects both ganglionic and aganglionic intestinal segments, and resembles other forms of enterocolitis. Pneumatosis intestinalis may be present. One occasionally sees abnormal submucosal blood vessels in HD. The abnormal arteries are most conspicuous in the transitional zone, where they appear thickened and may show bizarre microscopic changes. Adventitial fibromuscular dysplasia, as evidenced by increased collagen around the internal elastic lamina, marked hypertrophy of the media, and obliterative endarteritis, also develops (62). The vascular changes predispose to ischemic injury.
Histologic Variants
There are several histologic variants of HD. One associates with intestinal neuronal dysplasia. Generally, there is a hypoganglionic transitional zone at the cranial end of the aganglionic segment, but hyperganglionic segments can also be found.
Total colonic aganglionosis can be divided into two groups based on the histologic findings. Some cases are histologically similar to short segment and long segment disease, whereas in others, the bowel is aganglionic but there is little or no neural hyperplasia. The latter finding can lead to a false-negative diagnosis.
In HD with coexisting intestinal neuronal dysplasia (IND), the IND lies within, or just proximal to, the aganglionic
transitional zone. Patients have also been described with aganglionosis involving the entire colon and terminal ileum and coexisting jejunal and gastric IND. The IND accounts for residual symptoms in HD patients following pullthrough operations (63).
transitional zone. Patients have also been described with aganglionosis involving the entire colon and terminal ileum and coexisting jejunal and gastric IND. The IND accounts for residual symptoms in HD patients following pullthrough operations (63).
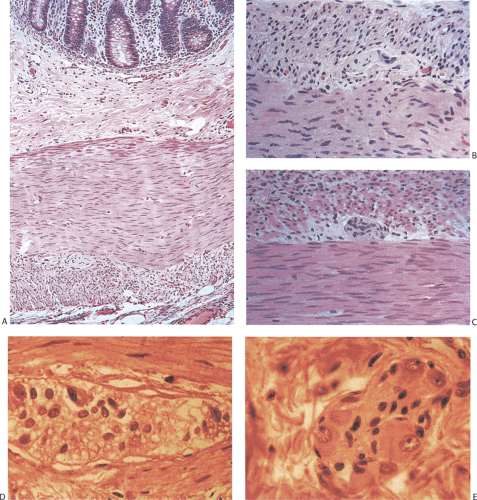 FIG. 10.11. Hirschsprung disease. A: Full-thickness section through the wall demonstrates the usual layers, but no ganglion cells. B: High-power magnification of the myenteric plexus shows abnormal small ganglion cell in the transitional area. C: A higher power magnification of another abnormal-appearing ganglion cell. D: Abnormal ganglion compared to a normal ganglion (E). |
Zonal aganglionosis, or skip segment HD, may affect some patients with total colonic aganglionosis (64). In this setting, one sees variable lengths of hypoganglionic or normoganglionic transverse or ascending colon resulting in a segmental aganglionic colon. Normal distal innervation or a skip
area containing some ganglia is present within an area of aganglionosis (64). Such cases are thought to have an ischemic origin. If biopsies are performed on the unaffected segments, the diagnosis will be missed.
area containing some ganglia is present within an area of aganglionosis (64). Such cases are thought to have an ischemic origin. If biopsies are performed on the unaffected segments, the diagnosis will be missed.
Special Techniques for Evaluating Hirschsprung Disease Specimens
Acetylcholinesterase (ACE) enzymatic staining (Fig. 10.12) demonstrates an increased network of coarse, thickened, irregular, cholinergic nerve fibers within the muscularis mucosae and lower mucosa. The lamina propria fibers travel in a plane parallel to the mucosal surface. Numerous submucosal small nerve fibers and smaller and larger nerve trunks may also be present. This pattern is evident even in the most distal biopsies, including those from the mucocutaneous junction. Increased ACE nerve fibers are consistently present in short and long segment HD, but they may be absent in total colonic aganglionosis. ACE staining patterns are also less dramatic in neonates than in older individuals, possibly leading to false-negative diagnoses.
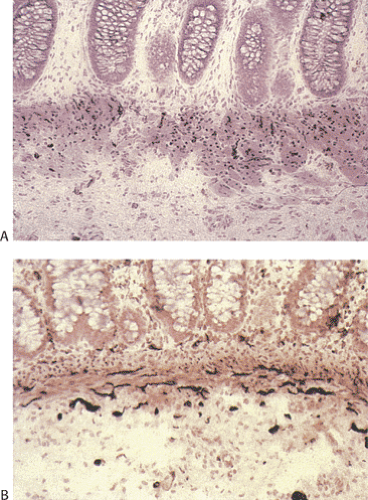 FIG. 10.12. Acetylcholinesterase staining reactions. The positive reaction is shown by the blackish brown color. A: Normal bowel characterized by the presence of thin, wispy, acetylcholinesterase-positive nerve fibers within the muscularis mucosa. None is present in the overlying lamina propria of the mucosa. B: Patient with Hirschsprung disease demonstrating thick, irregular fibers both within the muscularis mucosae and extending up into the lamina propria and around the glands. (Pictures courtesy of Dr. Kevin Bove, Cincinnati Children’s Hospital.) |
The common procedure for the ACE staining is as follows: Two mucosal suction biopsies are obtained 3 to 4 cm above the pectinate line. One is snap-frozen at the bedside. The other is placed in formalin for paraffin embedding. Delayed freezing after specimen delivery may cause false-negative results due to enzyme degradation. The frozen sample is cut in a plane perpendicular to the mucosal surface. Several paraffin sections are used to complement the evaluation of acetylcholinesterase activity. The special stains listed in Table 10.5 help delineate specific structures.
Treatment and Prognosis
Surgery is invariably necessary to treat symptomatic HD and the HD-associated enterocolitis. Persistent constipation is the most important long-term problem in patients operated on for HD. Inadequate resection, anastomotic strictures, coexisting IND, or achalasia of the internal anal sphincter may also cause these sequelae.
Intestinal Neuronal Dysplasia
IND, another developmental abnormality involving the ENS, occurs in two forms: Type A is characterized by decreased or immature gastrointestinal sympathetic innervation, and type B is characterized by increased numbers of ganglion cells, a dysplastic submucosal plexus, and defective neuronal nerve fiber differentiation (65,66). IND type A is also known as hypoganglionosis. IND type B is also known as hyperganglionosis. The entity known as oligoneuronal disease, which is sometimes called the hypogenetic type of dysganglionosis (67), may also be a form of intestinal neuronal dysplasia type A. These diseases are poorly understood and widely used consensus definitions for each entity are not available.
Intestinal Neuronal Dysplasia Type A
IND type A is very rare, and the symptoms caused by hypoganglionosis resemble those seen in HD. In newborns, there may be delayed meconium discharge; affected infants and small children have rare bowel evacuations that respond to enemas. With increasing age, fecal masses can be palpated through the abdominal wall. The colon becomes dilated and contains fecalomas. Distension causes intermittent colicky pain, often relieved by massive flatulence. Some children experience overflow discharge of sometimes bloody stool.
The diagnosis of hypoganglionosis is usually difficult to establish. X-ray studies, determination of transit times, and anorectal manometry are unreliable indicators of the disease.
The diagnosis of hypoganglionosis is usually difficult to establish. X-ray studies, determination of transit times, and anorectal manometry are unreliable indicators of the disease.
The disorder is characterized by an immaturity or hypoplasia of the extrinsic sympathetic nerves supplying the gut (68). Hypoganglionosis occurs in three forms: (a) an isolated form occurring as a segmental or even disseminated disease, (b) hypoganglionosis of variable length adjacent to an aganglionic HD, and (c) hypoganglionosis in combination with IND type B of a proximal segment. Hypoganglionosis may result from a developmental hypoplasia of the myenteric plexus (65), possibly due to the absence or abnormal expression of neurotrophic factors.
Patients with IND type A have reduced numbers of myenteric ganglia and myenteric plexus neurons, no or low colonic mucosal ACE levels, and secondary hypertrophy of the muscularis mucosae and the circular muscle of the muscularis propria. Absent or small submucosal and myenteric ganglia containing only one or two ganglia and immature neuroblastlike cells extend throughout the affected parts of the gut. Some patients have irreversible neuronal degeneration. Patients with IND type A may also have reduced numbers of ICCs, perhaps contributing to the dysmotility (69).
There is no consensus of how few ganglion cells there should be to make a diagnosis of hypoganglionosis. Meier-Ruge suggests that a 10-fold decrease in the number of ganglion cells per centimeter of bowel as compared to normal bowel is diagnostic (66). The distance between the ganglia in hypoganglionosis is nearly double that of the normal bowel. The treatment of hypoganglionosis (IND type A) is resection of the affected bowel and a pullthrough operation.
Intestinal Neuronal Dysplasia Type B
In contrast to IND type A, the incidence of IND type B varies from 0.3% to 40% of rectal suction biopsies (65,66). The mean age at diagnosis is 1.5 years. It occurs as an isolated disorder or it complicates many disorders (Table 10.10) (68,70). Patients with MEN-IIB and IND type B have RET mutations. In some cases, IND and neurofibromatosis are familial and associate with a tandem duplication in the NFI gene and a reciprocal translocation (t15;16) (q26.3;q12.1) (71). IND may also be one component of a complex malformation pattern.
IND type B clinically both mimics and complicates HD, as discussed in an earlier section. Patients present with nausea/vomiting, diarrhea, constipation, intestinal obstruction, intussusception, and volvulus. Symptoms develop insidiously, with progressive development of severe constipation that results in overflow incontinence (66). Many patients eventually spontaneously develop normal colonic motility (66). A significant number of patients develop severe intra-abdominal complications during the perinatal period, including necrotizing enterocolitis (NEC), meconium ileus, or bowel perforations. Such complications are especially common in premature neonates.
TABLE 10.10 Conditions That May Be Associated with Intestinal Neuronal Dysplasia | |
|---|---|
|
Neuronal dysplasia can be diffuse, involving both the small and large intestine, or it may remain confined to a single intestinal segment. Extensive disease may involve the stomach and esophagus. The bowel grossly appears either normal or variably dilated.
Controversy exists over the diagnostic criteria of IND type B. The diagnostic controversy is best exemplified by a study in which three pathologists agreed on the diagnosis in only 14% of children without aganglionosis (72). Smith also found that according to the criteria described by Borchard, only 11% of patients with megacolon had the obligatory criteria (hyperplasia of the submucosal plexus, an increase in ACE-positive nerve fibers around submucosal blood vessels, and ACE activity in the lamina propria) (73). The diagnosis of IND type B is also complicated by the fact that the density of ganglion cells in the myenteric plexus decreases significantly with age during the first 3 to 4 years of life and estimates of nerve cell density are influenced by section thickness (74).
The diagnostic criteria of IND type B have included prominent hyperplasia of the parasympathetic myenteric and submucosal plexuses characterized by increased numbers of neurons and ganglia (Fig. 10.13); giant submucosal ganglia containing 7 to 15 ganglion cells; hypertrophic nerve bundles containing increased numbers of thickened, beaded, and disorganized axons (Fig. 10.13); increased ACE activity
in mucosal, submucosal, and arterial adventitial nerves (66); a proliferation of fine nerve fibers in the lamina propria and circular muscle; and the presence of isolated ganglion cells in the submucosa, muscularis mucosae, or lower mucosa (Fig. 10.14) (65,66,67,68,72,73). The myenteric ganglia may be large and almost continuous with numerous readily identifiable neurons. The finding of giant ganglia, while almost always seen in IND type B, is not specific for it and this feature may be present in the proximal colon in patients with HD and in some patients with hypoganglionosis. Many neurons contain bizarre nuclei and poorly defined cytoplasm. Hypertrophic nerve bundles contain an increased number of thickened, beaded, and disorganized axons. These nerves may show an absence of NCAM and nicotinamide adenine dinucleotide phosphate (NADPH) diaphorase activity (75).
in mucosal, submucosal, and arterial adventitial nerves (66); a proliferation of fine nerve fibers in the lamina propria and circular muscle; and the presence of isolated ganglion cells in the submucosa, muscularis mucosae, or lower mucosa (Fig. 10.14) (65,66,67,68,72,73). The myenteric ganglia may be large and almost continuous with numerous readily identifiable neurons. The finding of giant ganglia, while almost always seen in IND type B, is not specific for it and this feature may be present in the proximal colon in patients with HD and in some patients with hypoganglionosis. Many neurons contain bizarre nuclei and poorly defined cytoplasm. Hypertrophic nerve bundles contain an increased number of thickened, beaded, and disorganized axons. These nerves may show an absence of NCAM and nicotinamide adenine dinucleotide phosphate (NADPH) diaphorase activity (75).
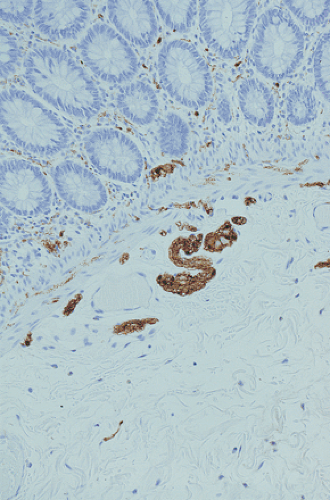 FIG. 10.13. Four-month-old infant with intestinal neuronal dysplasia type B. The hypertrophic nerves are accentuated by an S100 immunostain. |
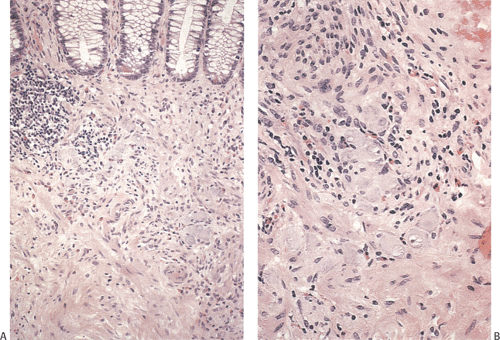 FIG. 10.14. Area of hyperganglionosis in the same specimen illustrated in Figure 10.13. A: Low magnification showing a large collection of ganglion cells lying in the hypertrophic muscle and in the lower portion of the mucosa. B: Higher magnification showing the large numbers of ganglion cells. |
Inflammatory changes, as might be seen in visceral myopathies or neuropathies, are absent. Prominent hypertrophy of the circular and longitudinal muscle layers also occurs.
Only about 5% of the ganglia in IND are giant ganglia (74). The specificity of giant ganglia as a marker for IND type B has been questioned, since occasional giant ganglia can be found in individuals without constipation. The presence of giant ganglia may be age-independent changes, whereas hyperplasia of the submucosal plexus and increases in ACE activity in the nerve fibers of the lamina propria appear to be age-dependent findings that disappear with maturation of the enteric nervous system. Therefore, neural
hyperplasia is significantly more common in neonates <4 weeks of age than in older individuals (76). This may explain our experience that one often sees hyperganglionosis in the absence of neural hyperplasia in young children with a history of chronic constipation.
hyperplasia is significantly more common in neonates <4 weeks of age than in older individuals (76). This may explain our experience that one often sees hyperganglionosis in the absence of neural hyperplasia in young children with a history of chronic constipation.
Patients with IND may also exhibit ICC hyperplasia (77). When it is marked, it can be visible grossly as a thick, white, fibrous band between the inner circular and outer longitudinal muscle layers throughout the full length of the resected bowel, especially in children with neurofibromatosis type 1 (NF-1). Microscopically, this bandlike layer consists of haphazardly arranged spindled to oval-shaped cells. The nuclei are long and oval in shape with slightly tapered ends and possess hyperchromatic or clumped chromatin and occasional small nucleoli. The cells have a moderate amount of eosinophilic cytoplasm; mitotic figures are rare. The muscle layers are partially replaced by these hyperplastic spindle cells and focally the full thickness of the inner muscular layer can be involved. Residual myenteric plexus can be identified in the midst of the hyperplastic cells. The hyperplastic cells are c-kit positive.
Patients with IND often have large numbers of mast cells in the bowel wall compared to the normal colon. Mast cells produce nerve growth factors that support the development and functional maintenance of the sympathetic and cholinergic neurons, and they may be important in the neuronal hyperplasia seen in this condition (78). We have also seen endocrine cell hyperplasia associated with hyperganglionosis in the neonate.
Individuals with IND also often have secondary changes in the muscularis propria. There may be areas of significant muscle atrophy in one or another layer of the muscularis propria. Alternatively, there may be hyperplasia of either the circular or longitudinal layer of the muscularis propria or these two changes may both be present. These changes may be focal or diffuse in nature and they may be present in the circular layer in some parts of the gut and in the longitudinal layer in others. These secondary changes undoubtedly reflect abnormal innervation of the muscle layers and the neuromuscular junction.
Overall, it would be desirable to have better quantitative diagnostic criteria of IND to distinguish normal variants from pathologic conditions, particularly in very young children. Moore et al introduced a morphologic scoring system based on the finding of hyperganglionosis, giant ganglia, neuronal maturity, heterotopic neuronal cells, and ACE activity in the lamina propria, muscularis mucosae, or adventitia of submucosal blood vessels. Hyperganglionosis and increased ACE activity of nerve fibers in the lamina propria had major importance in this scoring system (79). The best diagnostic indicator of IND in adults may be the detection of six to ten giant ganglia with more than seven nerve cells in 15 biopsy sections (80).
Some patients, especially those with IND type B, outgrow their disease as the ENS matures. Patients with persistent symptoms are managed medically with prokinetic agents, colonic irrigations, and cathartics. If bowel symptoms persist after 6 months of conservative treatment, surgery is often considered. Resection and pullthrough operations may be indicated in extensive IND.
Hyperganglionosis and Ganglioneuromatosis
Patients with hyperganglionosis and diffuse ganglioneuromatosis (Fig. 10.15) almost always have MEN-IIB and a mutation in the RET oncogene. The changes may associate with or be a form of IND type B.
Neuronal Immaturity
Neuronal immaturity, also known as neuronal maturational arrest syndrome, is characterized by the failure of neural elements to mature properly so that the ganglia appear immature and the patients present with clinical features resembling HD and IND. There may be overlap with IND. The underlying cause of failed neuronal maturation is unknown. Pathogenetic mechanisms may include (a) failure of normal numbers of neural crest cells to migrate into the gut, (b) inadequate neural proliferation in the gut, or (c) lack of growth or death of neuroblasts once they arrive in the gut due to the failure of the local microenvironment to support normal neuronal development during fetal life. There may also be a lack of neurotrophins since the latter are known to be important in neuronal development, differentiation maturation, survival, and maintenance. There may also be absence or delayed maturation of the ICCs, contributing to pseudo-obstruction in neonates.
The histologic features differ depending on the stage at which myenteric plexus development ceased. Patients exhibit several major histologic abnormalities: (a) no myenteric plexus seen in either H&E or specially stained sections, (b) small numbers of neuronal structures (ganglia and nerve trunks), or (c) an apparently normal myenteric plexus seen on H&E-stained sections but a neural deficiency shown by silver or immunohistochemical staining. The immature neurons lack neurofilaments. The ganglion cells may also line up at the periphery of the ganglia (ring-shaped ganglia) as occurs in premature infants (Figs. 10.16 and 10.17). There is no inflammation or neural degeneration. These findings contrast with those seen in patients with HD or IND type B in that there is no neural hyperplasia, but as noted there may be overlap with IND.
Neuronal immaturity often spontaneously improves with conservative therapy and the normal development of the child unless it occurs as part of IND. In other patients the changes persist.
Congenital Hyperplasia of Interstitial Cells of Cajal
As noted earlier, ICCs are the pacemaker cells of the GI tract that initiate peristalsis in the stomach and the intestines.
They are better known to pathologists as the cells of origin of gastrointestinal stromal tumors (see Chapter 19). These cells may be congenitally hyperplastic, presenting clinically as a pediatric motility disorder. A recent case of a 2-year-old girl was reported who had had scanty stool from birth. Radiographically, the distal colon was rigid and narrowed. The proximal colon was dilated and atonic. She underwent an ileocolectomy and the specimen showed a continuous proliferation of spindle cells located between the layers of the muscularis propria throughout the right colon. The spindle cells were positive for c-kit and CD34 but were negative for neural markers. In addition, the submucosa showed the changes characteristic of IND type B as discussed in an earlier section (77).
They are better known to pathologists as the cells of origin of gastrointestinal stromal tumors (see Chapter 19). These cells may be congenitally hyperplastic, presenting clinically as a pediatric motility disorder. A recent case of a 2-year-old girl was reported who had had scanty stool from birth. Radiographically, the distal colon was rigid and narrowed. The proximal colon was dilated and atonic. She underwent an ileocolectomy and the specimen showed a continuous proliferation of spindle cells located between the layers of the muscularis propria throughout the right colon. The spindle cells were positive for c-kit and CD34 but were negative for neural markers. In addition, the submucosa showed the changes characteristic of IND type B as discussed in an earlier section (77).
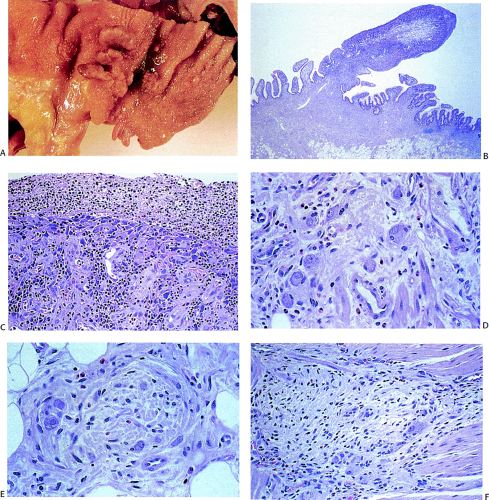 FIG. 10.15. Patient with pseudo-obstruction, intestinal neuronal dysplasia, and ganglioneuromatosis. A: Gross appearance of the bowel lumen demonstrates numerous polyps. B: Histologic examination shows a cellular proliferation that obliterates the normal mucosal–submucosal junction. C: Section through the base of the polyp showing a ganglioneuroma. D: Higher power magnification shows the neural tissue and ganglion cells. E: Submucosal ganglion demonstrating peripheral fibrosis and abnormal architecture. F: The architecture of the myenteric plexus is abnormal and there are numerous inflammatory cells within the myenteric plexus. (Case courtesy of Dr. E. Foucar, Albuquerque, NM.) |
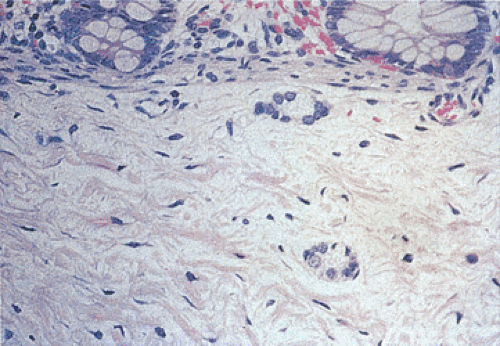 FIG. 10.16. Infant with pseudo-obstruction and neuronal immaturity. The submucosa contains ring-shaped ganglia. |
Absent Enteric Nervous System
The absence of nerves and ganglia from the stomach to the colon characterizes the absent enteric nervous system. This very rare disorder presents as severe perinatal pseudo-obstruction. No nerves or ganglia are present in either the submucosal or myenteric plexus, or in the muscularis propria. S100 and ACE staining confirm the absence of enteric neural structures. Sparse PGP 9.5–positive extrinsic nerve fibers are present in the submucosa. Prominent ICCs are present in the area between the circular and longitudinal muscle layers in a normal distribution in some parts of the gut. In other parts they are absent or in various stages of destruction. The overall prognosis is poor (81).
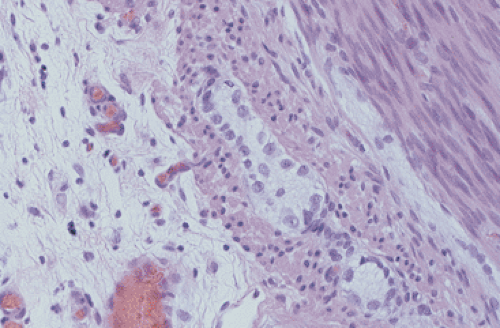 FIG. 10.17. Infant with pseudo-obstruction and neuronal immaturity. This picture of the myenteric plexus is from the same case that is shown in Figure 10.16. A number of immature ganglion cells line up at the periphery of the myenteric plexus. |
Pediatric Motility Disorders not Fitting Specific Diagnostic Criteria
Damage to the ENS results in enteric neuropathies, the best characterized of which is HD. However, a number of children present clinically with pseudo-obstruction syndromes in the first few weeks, months, or years of life who lack the classic features of either HD or IND (type A or B). These cases are challenging for clinicians to manage and they often look to the pathologist for help in establishing a diagnosis that will guide them into the appropriate therapy for the affected children. Pathologic features are not readily associated with clinical findings and vice versa.
The cases that we see in consultation are challenging in this regard since they often lack features that allow them to be easily categorized (Fig. 10.18). We have seen examples of neuronal immaturity with variable numbers of ganglia and with or without obvious neural disorganization in the bowel wall. Increased, thickened nerve fibers with or without giant ganglia or aganglionosis cases are also encountered. There may be cases with neural degeneration, variable inflammation, and increased apoptosis. We have also seen some cases associated with myenteric plexus loss and cases of hypo- or aganglionosis with shriveled ganglia and dense submucosal fibrosis that may be the equivalent of what has been described as zonal HD. Often in these cases there is a history of intestinal atresia, and we have interpreted such cases as representing prenatal neural injury rather than HD. This injury is likely to be ischemic in nature, complicating the complex series of intestinal twists and turns that associate with fetal intestinal development. Ischemia secondary to malrotational or other events may damage the neuromuscular structures of the gut, especially in the intestines.
The development of the gastrointestinal tract is in some ways more complex than other organ systems with marked lengthenings, rotations, and a cranial–caudal differentiation gradient. Additionally, as noted in earlier sections of this chapter, ENS development is a complex process that is governed by the interactions of numerous proteins that are responsible for neural migration, growth, differentiation, and survival. Alterations in any of these proteins can presumably result in disorders that present clinically like HD but fail to
meet the diagnostic criteria for that disease. Lastly, the developing ENS may be negatively influenced by other fetal events such as intrauterine infections that may lead to ganglionic loss or other neural abnormalities.
meet the diagnostic criteria for that disease. Lastly, the developing ENS may be negatively influenced by other fetal events such as intrauterine infections that may lead to ganglionic loss or other neural abnormalities.
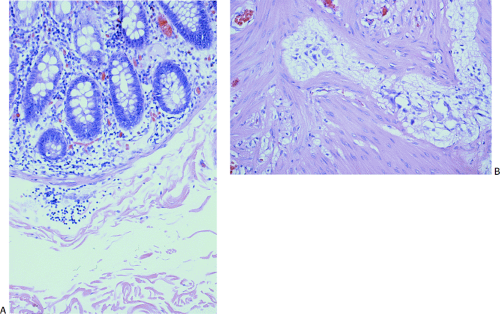 FIG. 10.18. Example of a neuropathy in a 1-month-old with pseudo-obstruction that is difficult to classify into currently recognized disorders. Ganglia are present in both the submucosal (A) and myenteric plexus (B). They are neither increased nor decreased in number but they appear small and shriveled. There is no neural hyperplasia.
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|





