Miscellaneous Tumors and Tumorlike Conditions
Ying-Bei Chen
RENAL CARCINOID TUMOR
Fewer than 100 cases of renal carcinoid tumor, a primary well-differentiated neuroendocrine neoplasm of the kidney, have been reported in literature (1,2,3,4,5,6,7,8). Although exceedingly rare, among the genitourinary organs, kidney is the second most common site for primary carcinoid tumors in both sexes after the gonads. About half of the patients are younger than 50 years of age at presentation. Females and males are equally affected. The presenting symptoms are usually back or flank pain, hematuria, or enlarging abdominal mass. Clinical signs or symptoms of carcinoid syndrome are very uncommon. Up to 20% of primary renal carcinoids arise in association with “horseshoe” kidneys (3). Rare tumor has also been reported to arise in renal teratoma or “teratoid tumors,” both being extremely uncommon tumors in the kidney (8). Extension of the tumors into the perinephric or renal pelvic fat is not uncommon. Regional lymph node or distant (e.g., liver, bone, lung) metastasis has been documented in more than one-third of the tumors. However, most patients with metastasis have protracted clinical course, with only rare reported death from the disease. Surgery remains the main treatment approach. The role of adjuvant chemotherapies or targeted therapies is still uncertain.
Renal carcinoid tumor is an exceptionally rare diagnosis rendered in kidney needle biopsies (9); however, it remains an important differential diagnostic consideration when tumors with neuroendocrine differentiation are encountered at this site. Morphologically, renal carcinoids are similar to carcinoid tumors at other sites. If present on a core biopsy, the junction of carcinoids and adjacent renal parenchyma is usually sharply defined (Fig. 7.1). The most commonly described architectural pattern is tightly packed cords and trabeculae with ribbonlike appearance (Figs. 7.2 and 7.3). Other growth patterns include solid sheets, solid nests, and the presence of gland-like lumina (Fig. 7.4). It is common to see a combination of architectural patterns within a tumor. Intervening stroma may vary from minimal to prominent with dense fibrotic bands.
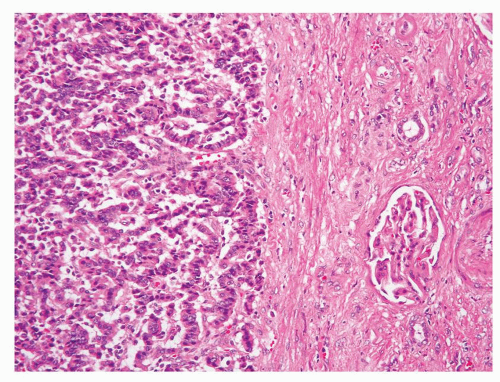 FIGURE 7.1 Renal carcinoid tumor often shows a sharp demarcation from the adjacent renal parenchyma. |
Like other well-differentiated neuroendocrine tumors, tumor cells are usually uniform and contain round or elongated nuclei with finely granular chromatin (“salt and pepper” appearance) and inconspicuous nucleoli. The nuclei are often situated perpendicular to the long axis of the cords or trabeculae. Focal, mild to moderate nuclear pleomorphism is
occasionally present. Calcifications, either focal or involving relatively large areas of the tumor, may be present in some cases. Rarely, metaplastic bone may be seen. Hemorrhage and cyst formation are also present in some cases. However, necrosis is extremely uncommon. Most renal carcinoid tumors exhibit less than 2 mitoses per 10 high-power fields (HPFs), but tumors can
rarely show 3 to 4 mitoses per 10 HPFs (Fig. 7.5). The relationship between renal carcinoid tumors and other rare cases of primary neuroendocrine tumors of kidney with high mitotic activity, such as small cell carcinoma and large cell neuroendocrine carcinoma, remains unclear.
occasionally present. Calcifications, either focal or involving relatively large areas of the tumor, may be present in some cases. Rarely, metaplastic bone may be seen. Hemorrhage and cyst formation are also present in some cases. However, necrosis is extremely uncommon. Most renal carcinoid tumors exhibit less than 2 mitoses per 10 high-power fields (HPFs), but tumors can
rarely show 3 to 4 mitoses per 10 HPFs (Fig. 7.5). The relationship between renal carcinoid tumors and other rare cases of primary neuroendocrine tumors of kidney with high mitotic activity, such as small cell carcinoma and large cell neuroendocrine carcinoma, remains unclear.
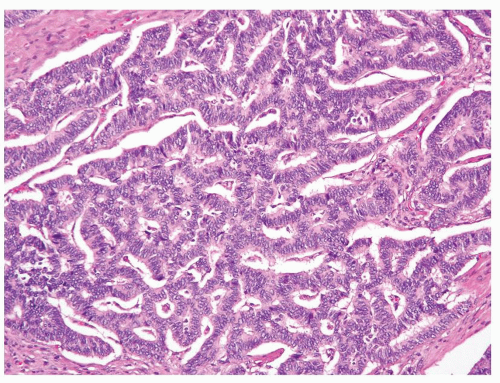 FIGURE 7.2 Ribbonlike tightly packed cords commonly seen in renal carcinoid tumor. |
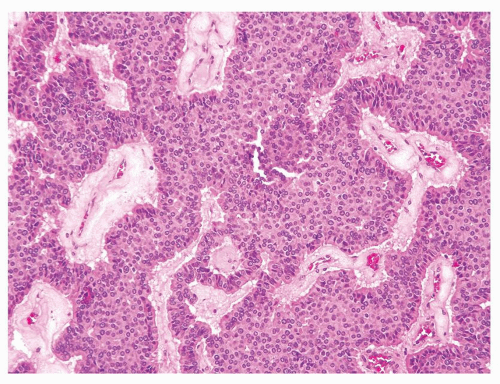 FIGURE 7.3 Anastomosing trabeculae in renal carcinoid tumor. |
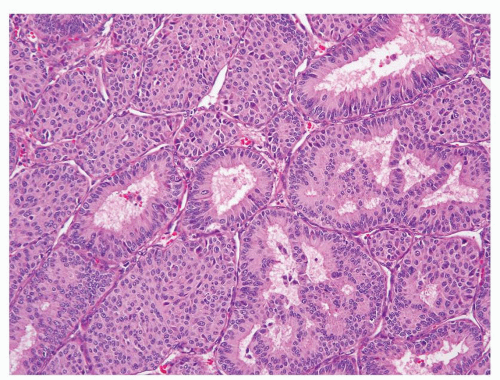 FIGURE 7.4 Carcinoid tumor with solid nests and gland-like lumina. |
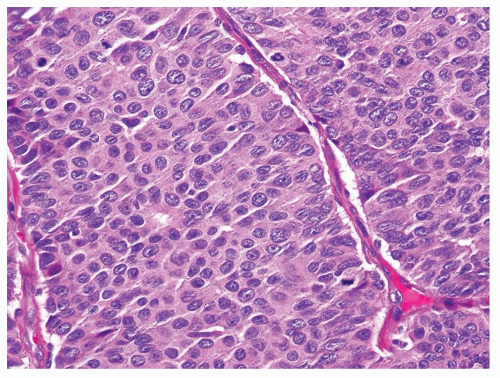 FIGURE 7.5 Carcinoid tumors can occasionally display increased mitotic activity. |
Ancillary Studies
Most of the renal carcinoids are positive for one or more neuroendocrine markers (synaptophysin, chromogranin, or CD56) (Fig. 7.6). Tumor cells also show variable reactivity to cytokeratins; Cam5.2 has the highest reported positivity at about 90% (1). In contrast to renal epithelial tumors, renal carcinoid tumors were found to be negative for PAX2 or PAX8 in a recent study (2). Whether this finding suggests a nonnephrogenic origin for renal carcinoids remains to be determined. It has also been recognized recently that some renal carcinoids show immunoreactivity to CD99, a feature that could lead to misdiagnosis of primitive neuroectodermal tumor (PNET) (2). Expression of TTF-1 has not been reported in renal carcinoids.
Differential Diagnosis
The differential diagnosis of primary renal carcinoid tumor mainly includes metastatic carcinoid tumors from other organ sites (lung, gastrointestinal [GI] tract, etc.) as well as other tumors with neuroendocrine features such as small cell carcinoma, PNET, or paraganglioma. It is noteworthy that chromophobe renal cell carcinoma (RCC) occasionally can contain areas morphologically mimicking a well-differentiated neuroendocrine tumor
(Fig. 7.7), which may lead to a misinterpretation as a renal carcinoid tumor on a core biopsy. However in contrast to carcinoids, these areas in chromophobe RCC are negative for neuroendocrine markers.
(Fig. 7.7), which may lead to a misinterpretation as a renal carcinoid tumor on a core biopsy. However in contrast to carcinoids, these areas in chromophobe RCC are negative for neuroendocrine markers.
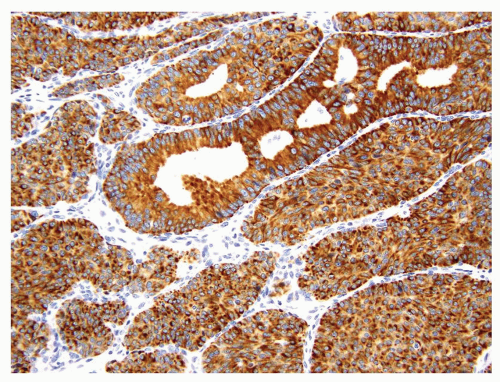 FIGURE 7.6 Strong immunoreactivity to synaptophysin often seen in renal carcinoids. |
The recognition of metastatic carcinoids relies mainly on clinical history. Metastatic carcinoids are also more likely to be multiple and are
often associated with lymphovascular tumor emboli. Small cell carcinoma demonstrates brisk mitotic activity, nuclear molding, and usually lacks the typical organized architectural patterns seen in carcinoids. PNET usually affects children and adolescents, although it may occur in adults. Given the reported CD99 positivity in renal carcinoids, histologic features and fluorescence in situ hybridization (FISH) testing of EWSR1 rearrangements would be very important to distinguish the two entities (see following discussion). Paraganglioma is also extremely rare in kidney. Tumor cells often have abundant granular cytoplasm, show nested growth pattern, and are surrounded by S100-positive sustentacular cells.
often associated with lymphovascular tumor emboli. Small cell carcinoma demonstrates brisk mitotic activity, nuclear molding, and usually lacks the typical organized architectural patterns seen in carcinoids. PNET usually affects children and adolescents, although it may occur in adults. Given the reported CD99 positivity in renal carcinoids, histologic features and fluorescence in situ hybridization (FISH) testing of EWSR1 rearrangements would be very important to distinguish the two entities (see following discussion). Paraganglioma is also extremely rare in kidney. Tumor cells often have abundant granular cytoplasm, show nested growth pattern, and are surrounded by S100-positive sustentacular cells.
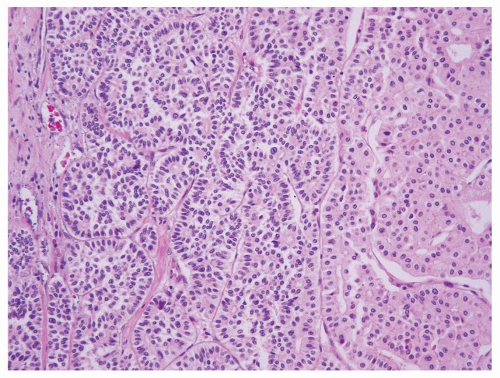 FIGURE 7.7 Chromophobe RCC may contain focal areas mimicking carcinoids (left). Note the adjacent area exhibiting features typical of chromophobe (right). |
PRIMITIVE NEUROECTODERMAL TUMORS
PNET is a rare entity in the kidney. Similar to its counterparts occurring in soft tissue and other sites, it is composed of monotonous, small, round primitive cells and characterized in almost all cases by recurrent translocations involving the EWSR1 gene on chromosome 22 and a member of the ETS family of transcription factors. These tumors typically infiltrate the surrounding renal parenchyma in broad sheets or finger-like projections (10). Most cases are composed of uniform small round cells with fine chromatin and scanty cytoplasm in a vaguely lobulated pattern (Fig. 7.8). Occasionally, histologic features suggesting neuroectodermal differentiation such as the formation of pseudorosettes can be found in some cases. High mitotic activity and necrotic areas are commonly present.
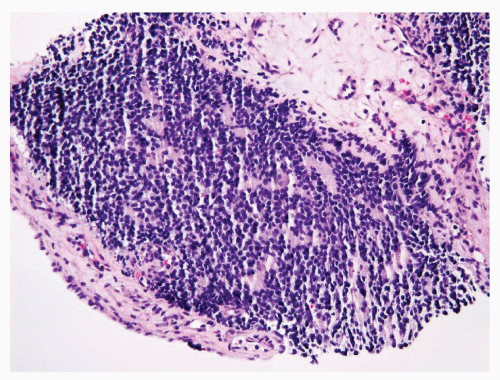 FIGURE 7.8 PNET detected in a core biopsy. |
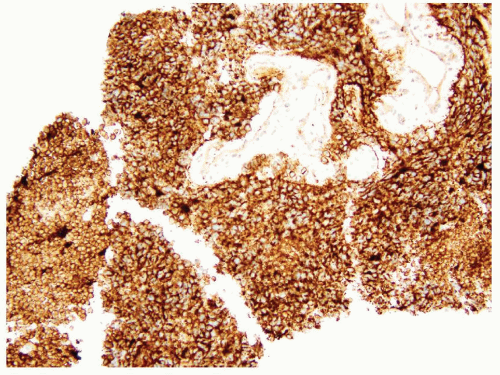 FIGURE 7.9 PNET shows diffuse membranous staining of CD99. |
Ancillary Studies
The vast majority of PNETs show strong and diffuse membranous immunoreactivity for CD99 (Fig. 7.9). Many cases, but not all, also display FLI-1 nuclear positivity. However, neither marker is entirely specific for the entity. For example, CD99 positivity can be seen in leukemic infiltration of soft tissues or extranodal lymphoma, therefore caution needs to be exercised when using these markers in core needle biopsies. Some PNETs can exhibit focal positivity for cytokeratins, but they are usually negative for desmin, skeletal muscle markers, and WT1. About 85% of all Ewing sarcoma/PNET harbor a somatic reciprocal chromosomal translocation, t(11;22)(q24;q12), that fuses EWSR1 to FLI1. Most of the remaining cases have alternative translocations between EWRS1 and other ETS family members (e.g., ERG, ETV1, etc.). FISH testing of EWSR1 rearrangements thus is a very helpful ancillary tool to confirm the diagnosis of PNET.
Differential Diagnosis
Renal PNETs need to be distinguished from other “round cell” tumors of the kidney, including blastema-predominant Wilms tumor, neuroblastoma, synovial sarcoma, desmoplastic round cell tumor, small cell carcinoma, lymphoma, or myeloid sarcoma. A combination of immunohistochemical and molecular studies usually can help establish a definitive diagnosis.
JUXTAGLOMERULAR CELL TUMOR (RENINOMA)
Juxtaglomerular cell tumor (JGCT) is a rare renin-secreting renal tumor that arises from juxtaglomerular apparatus (11,12,13,14,15). It usually occurs in young adults, with peak incidence in the second or third decades of life. But the reported age has ranged from 6 to older than 80 years old. It is seen more often in females than males (1.5:1 to 2:1). The characteristic clinical presentation is severe hypertension that shows minimal response to medical treatment. Patients often also have hyperaldosteronism and hyperkalemia, secondary to the high renin level. Surgical excision results in normalization of blood pressure in most cases, but some patients may remain hypertensive due to secondary angiopathy caused by the tumor and progress to renal failure. JGCT in general has been considered as a benign tumor. However, rare reported cases had metastasis to the lung (11).
Needle aspiration biopsies have been reported to show low sensitivity for JGCT (16). Often presenting as a unilateral, well-circumscribed tumor, its morphologic features can be highly variable. The typical tumor shows sheets of round or polygonal cells with central round nuclei containing inconspicuous nucleoli, clear to pale eosinophilic cytoplasm, and relatively distinct cell borders (Fig. 7.10). However, the tumor can also show spindle cells with indistinct cell borders that are present in sheets or irregular cords. Tumors usually exhibit a complex vascular pattern that mimic hemangiopericytoma (Fig. 7.11). Thickwalled hyalinized blood vessels are also common. Stromal component
ranges from scant to abundant, often with hyalinization or myxoid change. Dispersed lymphocytic infiltrates are also present. Entrapped renal tubules can be seen within some tumors; not uncommonly they appear to be hyperplastic and show papillary configuration (Figs. 7.12 and 7.13).
ranges from scant to abundant, often with hyalinization or myxoid change. Dispersed lymphocytic infiltrates are also present. Entrapped renal tubules can be seen within some tumors; not uncommonly they appear to be hyperplastic and show papillary configuration (Figs. 7.12 and 7.13).
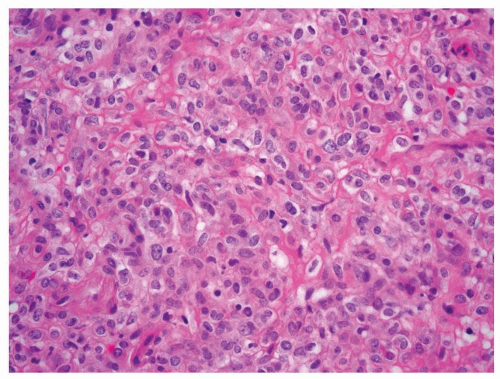 FIGURE 7.10 Juxtaglomerular cell tumor. |
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


