Mesenchymal Tumors
General Comments
Gastrointestinal (GI) spindle cell tumors caused diagnostic confusion for decades. Previously, most were diagnosed as either smooth muscle or neural tumors. However, following decades of ultrastructural and immunohistochemical studies, and more recently genetic investigations, it is now evident that most GI mesenchymal tumors arise from interstitial cells of Cajal (ICCs), the gastrointestinal pacemaker cells. For this reason, the term gastrointestinal pacemaker cell tumor (GIPACT) has been suggested (1). However, the more general term gastrointestinal stromal tumors (GISTs) has long been in vogue and has taken hold. While GISTs are the most common GI mesenchymal tumors, an array of other mesenchymal tumor types also develop in the GI tract.
Gastrointestinal Stromal Tumors
Demography
An estimated 5,000 to 6,000 new cases of GIST are diagnosed annually, with 10% to 30% of them being malignant (2). Most tumors are sporadic in nature, affecting individuals in their 5th or 6th decades of life. However, GISTs may arise in very young patients (3), and rare congenital tumors also exist (4). There is a slight male predominance. However, GISTs developing in the setting of the Carney triad (gastric GISTs, pulmonary chondromas, and extraadrenal paragangliomas) generally affect women under the age of 20 (5). GISTs developing in this setting are often multifocal, are purely epithelioid, and have a low risk of metastasis. GISTs arising in association with paragangliomas but without pulmonary chondromas may represent a variant of the Carney syndrome (5).
Familial GISTS develop in patients with germline-activating KIT (6) or platelet-derived growth factor receptor-α (PDGFRA) mutations (see below). Hyperpigmentation, mast cell tumors, and dysphagia are common to some kindreds (6). GISTS also develop in young patients with neurofibromatosis type 1 (NF1). The tumors are often multiple, usually developing in the small intestine (7). GISTs also complicate tuberous sclerosis (8) or following radiation therapy.
Cell of Origin
Most GISTs arise from CD117+ ICCs present in and around the myenteric plexus. ICCs show both myogenic and neural differentiation (1), explaining the immunohistochemical heterogeneity of the tumors that derive from them. Some GISTs originate from a CD34+ subset of ICC (9) or from primitive stem cells that can differentiate into ICC or smooth muscle cells (10).
Molecular Genetic Features
Constitutive activating KIT mutations occur in approximately 85% of GISTs. The KIT gene encodes a transmembrane tyrosine kinase receptor for stem cell factor (SCF). SCF receptor binding causes receptor dimerization and phosphorylation, inducing proliferation and inhibiting apoptosis (Fig. 19.1) (11). Activating mutations occur in exons 11 (21% to 71%), 9 (3% to 21%), 13, and 17 (Fig. 19.1) in a decreasing order of frequency (11,12,13,14). The type of KIT mutation is partially site and morphologically dependent (15,16,17). Spindle cell GISTs are most likely to contain exon 11 mutations, and exon 9 mutations most commonly affect intestinal GISTs (17). Novel germline mutations resulting in exon 8 deletions occur in familial GISTs and mastocytosis (18). KIT mutations range from single base pair substitutions to complex deletions/insertions. Internal tandem duplications may also be found (19). Generally, only one type of KIT mutation occurs in any given tumor. However, rare tumors contain two different somatic mutations. Recurrent tumors may show a different molecular phenotype than the primary tumor. So, for example, the primary tumor may show a mutation in exon 11 and the recurrence may contain both the exon 11 mutation as well as a new mutation in exon 13 (20).
Approximately 4% to 18% of GISTs harbor PDGFRA-activating mutations (17,21). The PDGFRA gene lies adjacent to the KIT locus and its structure and organization suggest that both genes derive from a common ancestral gene. PDGFRA mutations occur in exons 18, 12, and 14, with most involving exon 18 (16). Approximately 12% of GISTs lack KIT or PDGFRA mutations (16), indicating that there may be other rare, but yet unidentified genetic abnormalities that
contribute to GIST tumorigenesis. GISTs with KIT and PDGFRA mutations exhibit unique expression profiles and the genes identified by this technology may contribute to distinct clinicopathologic phenotypes. These gene products may also serve as highly selective therapeutic targets in GISTs containing KIT or PDGFRA mutations.
contribute to GIST tumorigenesis. GISTs with KIT and PDGFRA mutations exhibit unique expression profiles and the genes identified by this technology may contribute to distinct clinicopathologic phenotypes. These gene products may also serve as highly selective therapeutic targets in GISTs containing KIT or PDGFRA mutations.
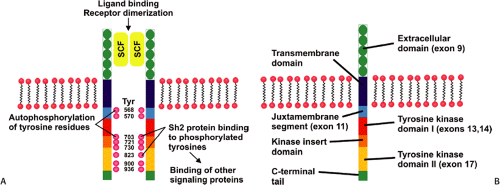 FIG. 19.1. Diagram of the kit receptor. A: Binding of stem cell factor to the receptor causes its dimerization, causing phosphorylation of tyrosine residues, which in turn activates downstream signals. B: The most common sites of mutation are illustrated. |
Familial GISTs associate with exon 8, 11, 13, and 17 KIT mutations (6,18) or mutations in the PDGFRA gene. In contrast, GISTs arising in the setting of NF1 tend to lack KIT or PDGFRA mutations (22). However, a novel NF1 mutation has been found in this setting (23). Nonfamilial GISTs associated with paragangliomas lack germline KIT mutations or mutations in the SDHA, SDHB, SDHC, and SDHD genes, genes typically associated with familial paraganglioma (24).
General Clinical Features
The clinical manifestations of GISTs vary depending on the site of origin, tumor size, and layer of the gut wall from which they arise. Many benign tumors remain asymptomatic, only to be found incidentally. However, they can become symptomatic, especially when large. Symptoms include dysphagia, abdominal pain, gastrointestinal bleeding, or obstruction. GI bleeding may present as chronic anemia or, occasionally, as massive hemorrhage. Some tumors may be palpated externally or during rectal examination.
General Gross Features
GISTs most commonly arise in the stomach (60% to 70%) followed by the small intestine (20% to 30%) (particularly the duodenum) (2), the colorectum (5%), and the esophagus (<5%). NF1 patients develop intestinal GISTs; gastric tumors are less common. Patients with NF1, familial GISTs, and the Carney triad often develop multiple GISTs.
GISTs usually center around the submucosa or muscularis propria. Most are well-circumscribed lesions surrounded by a thin pseudocapsule of compressed normal tissues. GISTs appear as single nodules, plaques, or multinodular lesions. Broad-based, intraluminal polypoid lesions may also be present. The tumors grow in an endocentric (Fig. 19.2) or exocentric fashion (Fig. 19.3). Tumors with both exocentric and endocentric growth patterns have a dumbbell shape (Fig. 19.4). The overlying mucosa can be intact or ulcerated. On cut section, GISTs lack the bulging, whorled cut surface characteristic of smooth muscle tumors. Instead, the cut surface appears pink and granular with patchy areas of hemorrhage, necrosis, or cystic degeneration (Fig. 19.5). Tumors vary in size from 0.5 to 45 cm, with a median size of 6 cm.
Interstitial Cells of Cajal Hyperplasia
Patients with familial GISTs or with NF1 often show a focal or diffuse ICC hyperplasia, particularly in the area of the myenteric plexus (Fig. 19.6). This change involves many regions of the gastrointestinal tract. The hyperplastic ICCs are polyclonal in nature (25) and are CD117+ and sometimes CD34+.
Common Histologic Features
Many GISTs are identified at the time of resection, when the diagnosis is established and a determination is made as to whether the tumor is benign or malignant. GISTs may also be diagnosed on biopsy (Fig. 19.7) or cytology specimens (Fig. 19.8). However, it is not realistic to attempt to distinguish a benign from a malignant GIST on biopsy or cytologic material unless the tumor is overtly malignant, since the features of malignancy are judged by many criteria in addition to the histologic features of the tumor (see below).
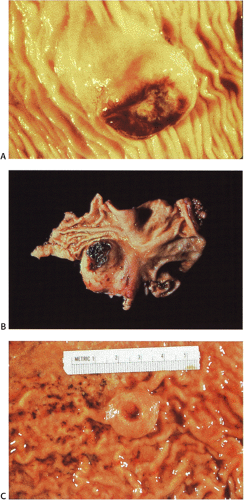 FIG. 19.2. Gastrointestinal stromal tumor (GIST) gross features. A: Small GIST with superficial ulceration and necrosis. The central ulcer is surrounded by a hyperemic surface consisting of granulation tissue. B: Large gastric leiomyoma with irregular superficial ulceration and a densely adherent clot on the surface. C: Small gastric GIST projecting into the lumen and producing a polypoid structure grossly resembling the nipplelike projections seen in some examples of heterotopic pancreas. At the time the stomach was removed for gross examination, it was filled with blood. |
GISTs can be divided into spindle cell, epithelioid, mixed, and pleomorphic lesions. Seventy percent of tumors are predominantly spindle cell in nature (2). The spindle cell components may exhibit a storiform, palisading, or herringbone pattern. The nuclei typically have blunt ends and are bullet or cigar shaped, but they can also be long and pointed. Some tumor cells have abundant cytoplasm; there may be areas of hyalinization and skenoid fibers. Epithelioid tumors consist of closely packed, polygonal cells. Some tumors also contain nests of small cells with an alveolar pattern. A small proportion of stromal tumors contain focal highly pleomorphic cells. These tumors usually have high mitotic rates measuring in excess of 10 mitoses/10 high-powered field (hpf).
 FIG. 19.3. Cut surface of a small intestinal gastrointestinal stromal tumor with a multilobulated irregular architecture. |
There is a wide clinical and pathologic spectrum ranging from indolent diminutive tumors to rapidly progressing sarcomas. Therefore, they have been extensively studied to better predict their biology, often with conflicting conclusions. As a result, a group of soft tissue pathologists met and developed consensus recommendations for assessing GIST biology (26) (Table 19.1). These generally work well, but they do not take into account tumor location, so one can add some refinements to these general recommendations based on tumor location, as discussed below.
The histologic features of the more common forms of GISTs are discussed by location. There are also rare histologic variants that occur anywhere in the gut. Gastrointestinal
autonomic nerve tumors (GANTs) are now known to represent a GIST variant. This variant is relatively more common in the small intestine and stomach and is relatively uncommon in the colon (27) and esophagus. The tumor contains small to intermediate-sized cells with a spindle or epithelioid appearance and a solid growth pattern. The cells exhibit long axonal processes containing cholinergic and adrenergic fibers and enveloping adjacent cells. Mitoses range from 1 to 23 mitoses/hpf (27,28).
autonomic nerve tumors (GANTs) are now known to represent a GIST variant. This variant is relatively more common in the small intestine and stomach and is relatively uncommon in the colon (27) and esophagus. The tumor contains small to intermediate-sized cells with a spindle or epithelioid appearance and a solid growth pattern. The cells exhibit long axonal processes containing cholinergic and adrenergic fibers and enveloping adjacent cells. Mitoses range from 1 to 23 mitoses/hpf (27,28).
 FIG. 19.4. Small intestinal gastrointestinal stromal tumor with dumbbell-shaped (exophytic and endophytic) growth pattern. |
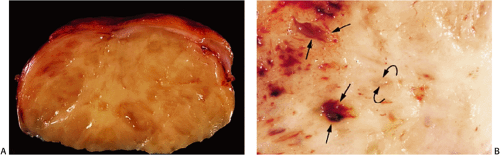 FIG. 19.5. Gastric gastrointestinal stromal tumor. A: Cut surface of the lesion. B: Higher magnification of the cut surface showing hemorrhagic cysts (arrows) and tan discolored areas (curved arrows) interspersed among tan-white solid areas. |
Another histologic GIST variant contains cells with signet ring cell features. These tumors frequently affect women and present as small (<2.5 cm), well-circumscribed, gastric, small intestinal, or rectal serosal nodules. Histologically, the lesions are characterized by a proliferation of large, round to oval cells containing abundant clear cytoplasm with nuclear displacement toward the cellular periphery (Fig. 19.9). These signet-appearing cells merge with more typical spindled cells. The tumor cells associate with a prominent myxoid matrix. Immunohistochemistry shows a heterogeneous staining pattern with strong positivity for vimentin and variable staining for CD34, S100, and actin. In the study initially describing these lesions (29), CD117 staining was not carried out since it predated the recognition that CD117 as a GIST marker.
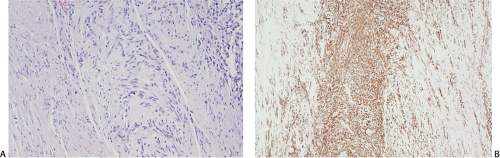 FIG. 19.6. Interstitial cells of Cajal hyperplasia. A: Histologically, this lesion appears as a neural hyperplasia in the area of the myenteric plexus. B: c-kit immunostain discloses the true nature of the lesion. |
The mesotheliomalike GIST variant typically contains epithelioid cordlike areas with pseudoglandular patterns distributed in a myxoid stroma (Fig. 19.10). GISTs with a rhabdoid phenotype (Fig. 19.11) contain paranuclear whorls of vimentin filaments (30). Oncocytic variants contain large numbers of mitochondria. Small cell variants contain cells with angulated nuclei that appear crowded together. Sometimes the tumor cells lie in tight perivascular whorls or balls of small spindle-shaped cells resembling paragangliomas. The cytotoxic T-lymphocyte–rich GIST (Fig. 19.12) arises in the stomach, esophagus, or mesentery, and resembles classic GISTs or GANTs with the exception that they are infiltrated by cytotoxic T cells (31). NF1-associated GISTs resemble non-NF1 spindle cell GISTs. Most contain skeinoid fibers and the tumors are typically surrounded by ICC hyperplasia. NF1-associated GISTs show dual differentiation with CD117+ ICCs and S100+ Schwann cells.
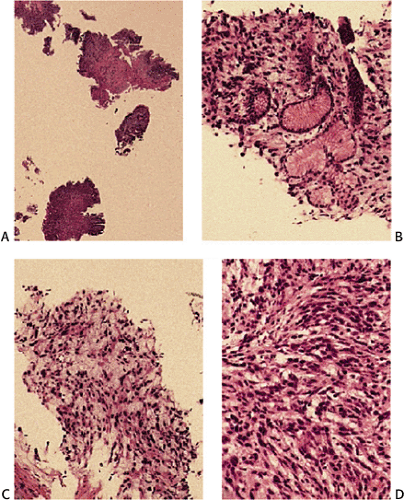 FIG. 19.7. Gastric gastrointestinal stromal tumor. A: This patient with known chronic active gastritis developed a mass lesion. Biopsy fragments of the most recent biopsy. The fragments differ in their appearance. The lower and middle biopsy fragments show evidence of chronic active gastritis. In the upper fragment, the gastric architecture is destroyed by the presence of an infiltrating mass. B: Higher magnification of an area of the upper biopsy fragment showing the replacement of the gastric mucosa by a cellular population with a high nuclear:cytoplasmic ratio that invades the lamina propria. This lesion can be diagnosed as malignant since there is mucosal invasion. C: Another area of the biopsy demonstrates a cellular neoplasm with a prominent myxoid stroma. D: This fragment is composed exclusively of spindle cells. |
Esophageal Gastrointestinal Stromal Tumors
Esophageal GISTs are rare and most are malignant. They present as intramural tumors or as polyps (32). They exhibit a cellular spindle cell pattern, or show areas of epithelioid differentiation. The histologic pattern varies, ranging from sheets of cells to areas with nuclear palisading and myxoid change. Some tumors demonstrate neural differentiation. These lesions are consistently CD34+ and c-kit positive with occasional actin immunoreactivity (32).
TABLE 19.1 Consensus Recommendations for Defining Risk of Aggressive Behavior in Gastrointestinal Stromal Tumors | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
Gastric Gastrointestinal Stromal Tumors
The stomach is the most common site of GISTs, where they are generally benign, but this is influenced by the features listed in Table 19.1 and by gastric location. There is a high frequency of malignancy in fundic and gastroesophageal GISTs, as compared with antral GISTs. Gastric GISTs show
two morphologic phenotypes that are relatively specific. One is a cellular spindle cell stromal tumor characterized by fascicles of spindle cells, often with pronounced palisades, monotonous and uniform nuclei, and perinuclear vacuoles, which indent the nucleus. Occasional large nuclei may occur. Hyalinization and myxoid degeneration are common. Mitotic activity is low. Epithelioid GISTs constitute most of the remaining GISTs. The tumors contain round epithelioid cells with prominent clear cytoplasm and cytoplasmic perinuclear vacuolization. The tumor cells lie in sheets or packets rather than in fascicles; they tend to be oriented in a perivascular pattern. In the stomach, most epithelioid GISTs are benign, provided mitotic counts do not exceed five mitoses per 50 hpf (33).
two morphologic phenotypes that are relatively specific. One is a cellular spindle cell stromal tumor characterized by fascicles of spindle cells, often with pronounced palisades, monotonous and uniform nuclei, and perinuclear vacuoles, which indent the nucleus. Occasional large nuclei may occur. Hyalinization and myxoid degeneration are common. Mitotic activity is low. Epithelioid GISTs constitute most of the remaining GISTs. The tumors contain round epithelioid cells with prominent clear cytoplasm and cytoplasmic perinuclear vacuolization. The tumor cells lie in sheets or packets rather than in fascicles; they tend to be oriented in a perivascular pattern. In the stomach, most epithelioid GISTs are benign, provided mitotic counts do not exceed five mitoses per 50 hpf (33).
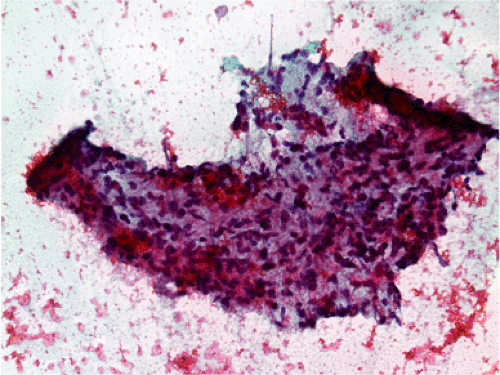 FIG. 19.8. Fine needle aspirate of a gastric gastrointestinal stromal tumor. A highly cellular tissue fragment is present that consists predominantly of spindle cells. There are no mitoses. |
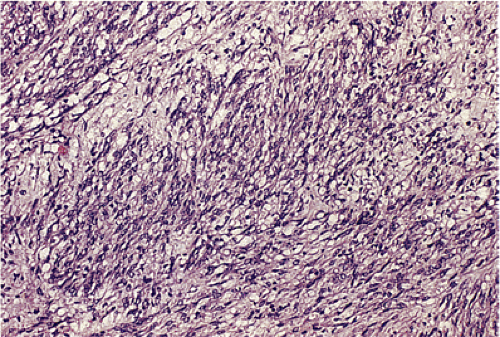 FIG. 19.9. Gastrointestinal stromal tumor with signet ring features. |
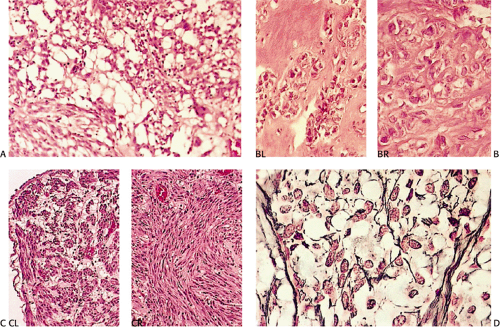 FIG. 19.10. Small intestinal gastrointestinal stromal tumor that presented in an inguinal hernia and had a pattern reminiscent of a mesothelioma. The photos are from the primary lesion in the ileum. A: Note the mixture of spindle cells and epithelioid areas. B: Ileal serosal surface showing epithelioid nests (left). Higher magnification (right). C: Other areas of the primary tumor. Epithelioid cells are seen on the left and spindle cells on the right. D: Reticulin stain demonstrating absence of reticulin. |
There is no consensus on the appropriate classification of gastric GISTs. One can follow the consensus recommendations discussed earlier or report gastric GISTs, noting the maximum mitotic rate and maximum tumor diameter and making a comment about the adequacy of the resection margin, tumor cellularity, and the presence or absence of mucosal invasion. Alternatively, one can use one of the two classifications described below.
Trupiano et al divided gastric GISTs into benign and malignant spindle cell GISTs, benign and malignant epithelioid GISTs, and benign and malignant mixed lesions (34). The predominant cell type is determined by the presence of >75% of the tumor being represented by either spindle cells
or epithelioid cells. Tumors that do not meet these criteria are classified as mixed tumors. Nuclear grade is considered to be high if the nuclei are large with irregular nuclear membranes and vesicular chromatin. Tumors without these features were classified as low grade (34).
or epithelioid cells. Tumors that do not meet these criteria are classified as mixed tumors. Nuclear grade is considered to be high if the nuclei are large with irregular nuclear membranes and vesicular chromatin. Tumors without these features were classified as low grade (34).
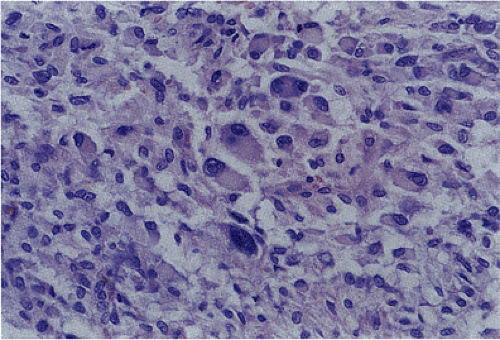 FIG. 19.11. Gastric gastrointestinal stromal tumor with a rhabdoid phenotype. |
According to this classification, benign cellular spindle cell GISTs are highly cellular tumors, with uniform spindle cells with an abundant pale to eosinophilic cytoplasm. The compact cells exhibit a patternless, fascicular, whorled, storiform, or palisading architecture. The uniform pale nuclei contain evenly distributed chromatin, inconspicuous nucleoli, and regular nuclear borders (Fig. 19.13). Mitotic activity is typically less than or equal to two figures per 50 hpf. Perinuclear vacuoles may be present. The tumor cells are often separated by a hyalinized or calcified stroma. There may be areas of liquefactive necrosis with pools of acellular material separating perivascular tumor islands (34).
Benign epithelioid GISTS, the most common gastric GISTs, consist predominantly, or exclusively, of epithelioid cells, often with well-defined borders, arranged in nests or sheets (Fig. 19.14). The cells have abundant cytoplasm that may be eosinophilic, amphophilic, or clear. There is often a condensed rim of eosinophilic cytoplasm adjacent to the nucleus with peripheral cytoplasmic clearing that is only appreciated on hematoxylin and eosin (H&E) examination, due to the fact that this is a fixation-induced artifact (Fig. 19.15). The nuclei are usually round with small nucleoli, but scattered multinucleated giant cells or cells with bizarre nuclei can be present. Mitotic figures are rare (usually two or less per 50 hpf). Stromal alterations include hyalinization and calcification (34). A rich vascular supply can cause a neuroendocrine or glomus appearance.
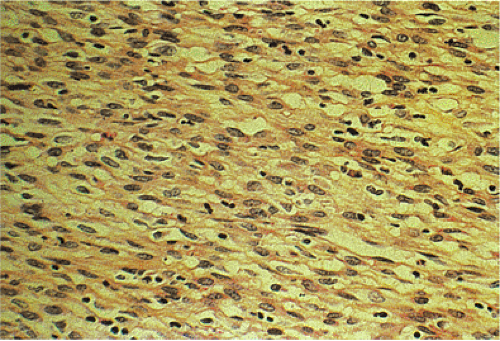 FIG. 19.12. Gastrointestinal stromal tumor with scattered cytotoxic T cells. |
In contrast to benign cellular GISTs, malignant spindle cell GISTs are larger, more cellular tumors containing cells with a high nuclear:cytoplasmic ratio (Fig. 19.16). The cytoplasm may appear eosinophilic, basophilic, or amphophilic. The nuclei vary in size and often appear vesicular. The perinuclear vacuoles typical of benign lesions are often absent; areas of tumor necrosis are common. Individual tumor cells may be arranged in storiform or fascicular patterns. Some tumors
show prominent nuclear palisading. Mitoses often number >10 mitoses/50 hpf. Mucosal invasion, as defined by infiltration of tumor cells across the muscularis mucosae between the glands at the base of the mucosa, may be seen (Fig. 19.7).
show prominent nuclear palisading. Mitoses often number >10 mitoses/50 hpf. Mucosal invasion, as defined by infiltration of tumor cells across the muscularis mucosae between the glands at the base of the mucosa, may be seen (Fig. 19.7).
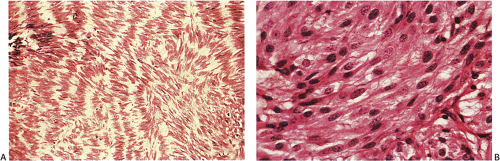 FIG. 19.13. Gastric gastrointestinal stromal tumor. A: Foci of spindled cell palisades. B: Higher magnification of the spindle cells. |
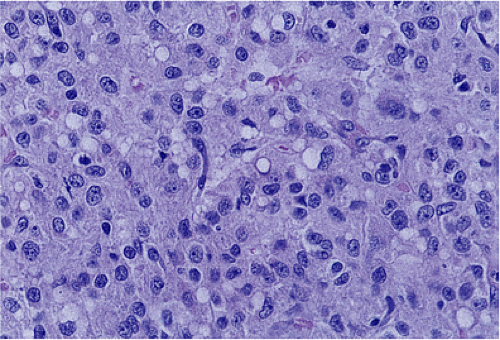 FIG. 19.14. Epithelioid gastrointestinal stromal tumor. The tumor consists of large polygonal cells. |
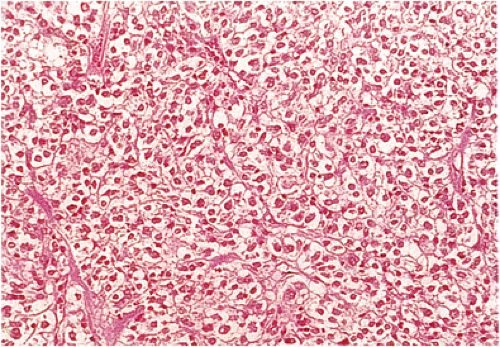 FIG. 19.15. Epithelioid gastrointestinal stromal tumor with so-called “fried egg” appearance. |
Malignant epithelioid GISTs consist of densely packed cells with less cytoplasm than the spindle cell variant. They are more cellular than their benign counterparts. The cells may be arranged in small acinarlike clusters or large sheets. A prominent myxoid stroma is often present. The nuclei are frequently hyperchromatic and monotonous in appearance. Some cells are pleomorphic. However, scattered bizarre cells are more common in benign epithelioid GISTs. Since the mitotic activity overlaps with that seen in benign epithelioid tumors (unless the mitoses are numerous), mitoses cannot be used to separate benign from malignant lesions. Furthermore, because benign-appearing areas are often present in malignant epithelial GISTs, extensive sampling is required to identify the malignant component (34).
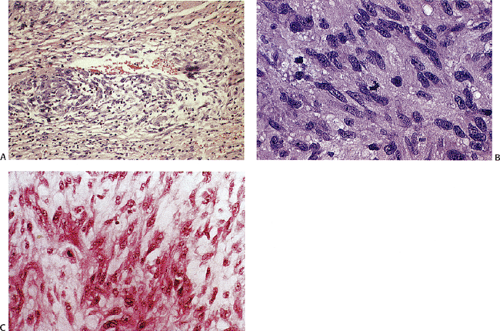 FIG. 19.16. Malignant spindle cell gastrointestinal stromal tumor. A: Low-power view showing atypical cells and focal necrosis. B: Medium-power view showing moderate cellular atypia and increased mitotic activity. C: Immature spindled and stellate cells with increased mitotic activity are present in a myxoid stroma. |
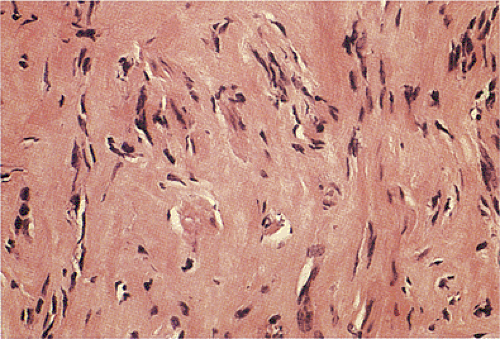 FIG. 19.17. Sclerosing spindle cell gastrointestinal stromal tumor. |
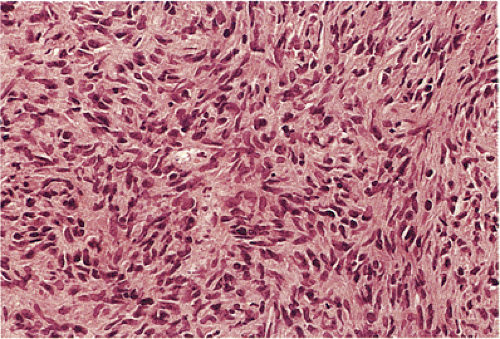 FIG. 19.18. Sclerosing epithelioid gastrointestinal stromal tumor. The figure demonstrates the presence of a densely cellular lesion with crowded nuclei. No mitotic activity is present. This area was surrounded by more typical spindle-shaped cells. |
A recent large study of gastric GISTs (765 cases with long-term follow-up) delineated eight histologic subtypes of gastric GISTs, an admixture of the subtypes, and a group of tumors that were unclassified (8). The eight types are as follows:
Sclerosing spindle cell GISTs are paucicellular tumors that show extensive extracellular collagen (Fig. 19.17) spindle cells, no nuclear atypia, low mitotic activity, and common calcification. They are usually small, although 13% were >10 cm.
Palisading and vacuolated spindle cell GISTs (Fig. 19.13) consist of cellular, plump, uniform spindle cells with nuclear palisading, perinuclear vacuolization, and limited atypia; mitotic activity rarely exceeded 10 mitoses/50 hpf.
Hypercellular spindle cell GISTs contain uniformly densely packed cells with diffuse sheets of spindle cells exhibiting limited atypia, nuclear palisading, and perinuclear vacuolization. The mitotic activity rarely exceeded 15 mitoses/50 hpf.
Sarcomatous spindle cell GISTs (Fig. 19.16) contain spindle or oval cells with diffuse atypia, and the tumor cells were often in bundles separated by a myxoid stroma. The mitotic activity is >20 mitoses/50 hpf; nearly all tumors are >5 cm in diameter.
Sclerosing epithelioid GISTs (Fig. 19.18) exhibit a syncytial pattern and consist of cohesive uniform polygonal cells with indistinct cell borders and diffuse collagenous matrix. Multinucleation and low mitotic rate are characteristic.
Epithelioid GISTs with discohesive patterns (Fig. 19.19) consist of large polygonal cells with abundant cytoplasm, distinct cellular borders, discohesive growth patterns, scant interstitial matrix, multinucleation, possible focal atypia, and low mitotic rates.
Hypercellular epithelioid GISTs (Fig. 19.20) contain back-to-back epithelioid cells with well-defined borders, nuclear atypia, and a higher nuclear:cytoplasmic ratio than the epithelioid GISTs with discohesive patterns. Mitotic activity rarely was >10 mitoses/50 hpf.
Sarcomatous epithelioid GISTs contain epithelioid to rounded cells with well-defined borders, high nuclear:cytoplasmic ratios, uniform nuclei, prominent nucleoli, and conspicuous mitotic activity (>20 mitoses/50 hpf).
The tumors are also divided into eight groups, based on maximum tumor diameter and mitotic activity (Table 19.2)
(8). The spectrum from the sclerosing, palisading–vacuolated, hypercellular, to sarcomatous among spindle cell GISTs reflected increasing frequency of adverse outcome. The sarcomatous type differed significantly from the others in terms of tumor-specific survival. The epithelioid sarcomatous tumors had a slightly better prognosis than analogous spindle cell tumors. Myxoid changes may be seen in both spindle cell and epithelioid cell lesions (Fig. 19.21).
(8). The spectrum from the sclerosing, palisading–vacuolated, hypercellular, to sarcomatous among spindle cell GISTs reflected increasing frequency of adverse outcome. The sarcomatous type differed significantly from the others in terms of tumor-specific survival. The epithelioid sarcomatous tumors had a slightly better prognosis than analogous spindle cell tumors. Myxoid changes may be seen in both spindle cell and epithelioid cell lesions (Fig. 19.21).
TABLE 19.2 Suggested Guidelines for Assessing the Malignant Potential of Gastric Gastrointestinal Stromal Tumors of Different Sizes and Mitotic Activity | ||
|---|---|---|
|
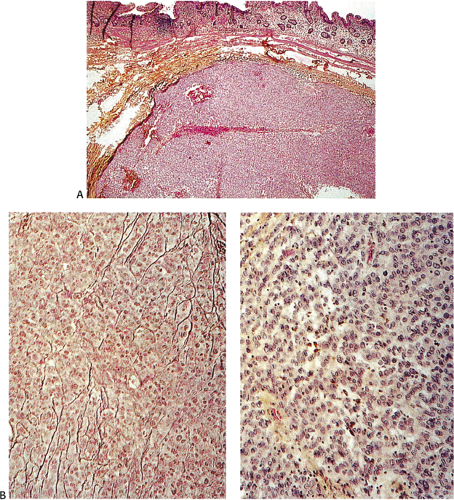 FIG. 19.19. Gastrointestinal stromal tumor (GIST) with discohesive cells. A: GIST arising in submucosa with a pushing margin extending to the muscularis mucosae. B: Composite picture of characteristic epithelioid to oval cells (right) and reticulin stain (left) circumscribing individual cells and groups of cells. |
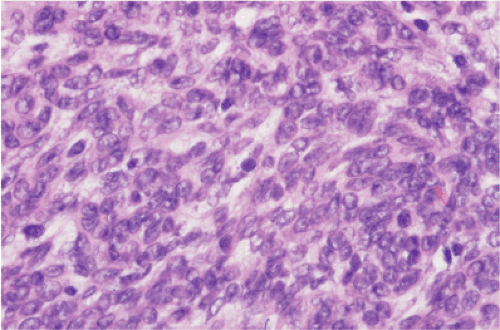 FIG. 19.20. Hypercellular epithelioid gastrointestinal stromal tumor. The mitotic activity in this cellular lesion with overlapping nuclear borders was three mitoses per 50 hpf. |
Features that appear to correlate with an aggressive clinical behavior of gastric GISTs include tumor mitotic activity, maximum tumor diameter, high nuclear grade, high cellularity, mixed cell type, mucosal invasion, tumor cell necrosis, and stromal alterations such as extensive myxoid change and absence of hyalinization (34).
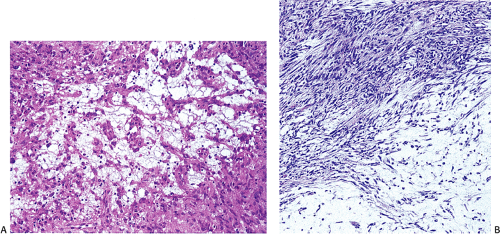 FIG. 19.21. Myxoid changes in gastrointestinal stromal tumor (GISTs). A: Focal area of myxoid degeneration showing abundant pale, amphophilic mucoid stroma. B: Another lesion demonstrating the presence of typical spindled cells of the GIST and adjacent myxoid areas. |
Small Intestinal Gastrointestinal Stromal Tumors
As a group, a larger percentage of small bowel GISTs are malignant when compared to gastric GISTs. The classic benign small intestinal GIST is a small lesion (measuring <5 cm) that consists of a uniform population of cytologically bland spindle cells with abundant eosinophilic cytoplasm and overall low tumor cellularity. The cells are usually distributed in nests separated by fine fibrovascular septae producing an organoid growth pattern reminiscent of paragangliomas. Eosinophilic collagen globules (skeinoid fibers) are characteristic and are often numerous, especially in duodenal lesions (Fig. 19.22). These fibers are most common in smaller, mitotically less active tumors. Focal calcification may be present. Nuclear palisading suggestive of neural tumors occurs in many tumors. The tumors may also contain distinctive hemangiomalike, sometimes glomerularlike vascular proliferations lying between the sheets of tumor cells. Perivascular
hyalinization is also common (33). Mitoses are low (less than five per 50 hpf). Benign tumors lack tumor cell necrosis and mucosal invasion (35).
hyalinization is also common (33). Mitoses are low (less than five per 50 hpf). Benign tumors lack tumor cell necrosis and mucosal invasion (35).
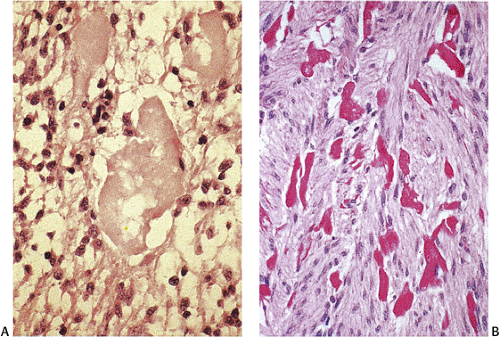 FIG. 19.22. Small intestinal gastrointestinal stromal tumor with skeinoid fibers. A: Hematoxylin and eosin section demonstrating the presence of amorphous aggregates of eosinophilic material typical of skeinoid fibers within the tumor. The tumor cells themselves are round and epithelioidlike in appearance or more elongated. B: Periodic acid–Schiff (PAS) stain demonstrating the PAS positivity of the material. |
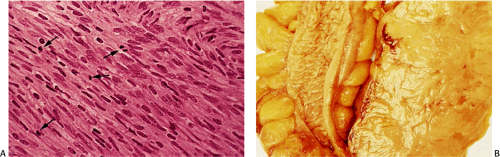 FIG. 19.23. Malignant small intestinal gastrointestinal stromal tumor. A: Low-power photograph showing the presence of a cellular spindle cell lesion with numerous mitoses (arrows). B: Gross features of the lesion shown in A. A mass surrounds the bowel. |
Most malignant small intestinal GISTs consist of highly cellular spindle cell proliferations (Fig. 19.23) with more cytologic atypia than occurs in benign tumors. The nuclei are larger with more coarsely clumped chromatin; mitoses are easily identified (greater than five per 50 hpf). The cells are typically arranged in long fascicles, as opposed to the organoid pattern seen in benign tumors. Skeinoid fibers are few in number or absent, and many, but not all, malignant small intestinal GISTs have tumor cell necrosis and/or mucosal invasion. Tumors with a conspicuous epithelioid component comprising more than 25% of the tumor are virtually always malignant (24,36,37). Since many malignant small intestinal GISTs contain benign areas, as well as malignant areas, these tumors should be well sampled to detect the malignant areas.
In one study, small intestinal GISTs were divided into the six categories shown in Table 19.3. Significant prognostic features included tumor size (>5 cm), mitoses (greater than five per 50 hpf), the presence of coagulation necrosis, an epithelioid histology, the absence of hyalinized vessels and skeinoid fibers, and the presence of mucosal invasion (33,38).
TABLE 19.3 Miettinen Classification of Small Intestinal Gastrointestinal Stromal Tumors | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||
Appendiceal and Large Intestinal Gastrointestinal Stromal Tumors
Appendiceal GISTs are spindle cell tumors that may contain skeinoid fibers. They lack atypia and mitotic activity (39). Colorectal GISTs are almost always spindle cell tumors, of which 50% are malignant, often with a highly aggressive clinical course. An infiltrative border, greater than 5 mitoses per 50 hpf (40), and mucosal invasion (Fig. 19.24) correlate with metastasis or death. The prognostic impact of tumor size is more controversial than in tumors arising in other locations (40). Colonic GISTs that contain skeinoid fibers have a better prognosis than those without them. Occasionally, malignant large intestinal GISTs contain osteoclastlike giant cells (40).
Anorectal GISTs are rare; most arise within the muscularis propria (41). They are generally a homogeneous group of cellular tumors composed predominantly of spindle cells; skeinoid fibers are absent. Small submucosal lesions without mitoses and pleomorphism behave in a benign fashion;
malignant tumors often have an infiltrative growth pattern and mitotic rates of five or more mitoses per 50 hpf. The impact of nuclear pleomorphism, necrosis, mitotic count, and intramuscular location is currently unclear (42). These tumors require long-term follow-up to determine their behavior. Deep intramuscular, cellular tumors should be considered to be malignant if they are >5 cm in diameter, infiltrate the muscularis propria, have one or more mitotic figure per 50 hpf, or contain coagulative necrosis or pleomorphism (42).
malignant tumors often have an infiltrative growth pattern and mitotic rates of five or more mitoses per 50 hpf. The impact of nuclear pleomorphism, necrosis, mitotic count, and intramuscular location is currently unclear (42). These tumors require long-term follow-up to determine their behavior. Deep intramuscular, cellular tumors should be considered to be malignant if they are >5 cm in diameter, infiltrate the muscularis propria, have one or more mitotic figure per 50 hpf, or contain coagulative necrosis or pleomorphism (42).
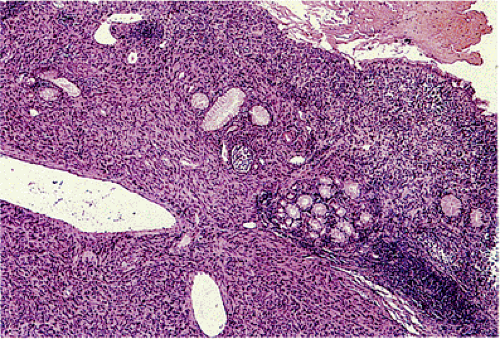 FIG. 19.24. Intestinal malignant gastrointestinal stromal tumor. The tumor diffusely invades the mucosa. Residual trapped intestinal glands are evident. A band of chronic inflammation is associated with some of the residual glands. |
Histologic Features of Gastrointestinal Stromal Tumors Following Treatment
Treated tumors may change their histology following treatment. Spindle cell tumors may develop an epithelioid or pseudopapillary epithelioid growth pattern characterized by rounded cells with an eosinophilic cytoplasm and uniform round to ovoid nuclei. There is a dramatic decrease in tumor cell density with the formation of a myxohyaline stroma, extensive hemorrhage, necrosis, and cystic transformation. There is usually a marked decrease in its proliferative rate (43). The tumor may lose its CD117 positivity with only a few residual positive cells (44). Some tumors also become CD34 negative. Some tumors become desmin positive (45).
TABLE 19.4 c-kit–expressing Tumors | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||
Immunohistochemical Features of Gastrointestinal Stromal Tumors
The definitive marker for a GIST (benign and malignant) is c-kit protein (CD117) expression (26,46). However, c-kit immunoreactivity can be seen in other tumors (Table 19.4) and not all GISTs are positive. The interpretation of the staining patterns is complicated by the fact that there are several commercially available antibodies and variable staining methodologies may result in false-positive and false-negative results. Additionally, CD117 positivity may be lost in treated tumors. Efforts are currently under way to validate c-kit staining. Further, CD117 immunohistochemistry tissue microarrays are available that facilitate interlaboratory comparisons of staining results, quality assurance programs, and educational purposes (47).
The predominant CD117 staining pattern is a diffuse cytoplasmic positivity (Fig. 19.25). Membrane staining or dotlike positivity may also occur. Rarely, CD117 positivity is present in only a portion of cells. PDGFRA immunostaining can be helpful in discriminating between c-kit–negative GISTs and other mesenchymal lesions. Other mesenchymal tumors are PDGFRA negative except for a subset of desmoid tumors (48).
Approximately 70% of GISTs express CD34 (Fig. 19.26) (48). CD34 immunoreactivity varies from 47% in small intestinal GISTs to 96% to 100% in rectal and esophageal tumors. Absence of CD34 expression in benign GISTs may indicate that they arise from more mature ICCs, whereas malignant GISTs may contain dedifferentiated ICCs that also express CD34 (49).
GISTs are variably positive with both smooth muscle (Fig. 19.27) and neural markers. Smooth muscle actin expression is most common in small intestinal GISTs (47%) and most rare in rectal and esophageal GISTs (10% to 13%). When
actin is expressed, it is generally focal, indicating that smooth muscle differentiation is incomplete. Heavy caldesmon, an actin-binding cytoskeleton-associated protein, is also expressed in some GISTs. Desmin staining is less common and is typically limited to scattered tumor cells, with more prominent staining in epithelioid tumors. S100 protein expression occurs in 10% to 15% of GISTs, usually in small intestinal GISTs, in a focal staining pattern. Scattered tumor cells may be cytokeratin positive. Eighty percent of GISTs stain with BCL2.
actin is expressed, it is generally focal, indicating that smooth muscle differentiation is incomplete. Heavy caldesmon, an actin-binding cytoskeleton-associated protein, is also expressed in some GISTs. Desmin staining is less common and is typically limited to scattered tumor cells, with more prominent staining in epithelioid tumors. S100 protein expression occurs in 10% to 15% of GISTs, usually in small intestinal GISTs, in a focal staining pattern. Scattered tumor cells may be cytokeratin positive. Eighty percent of GISTs stain with BCL2.
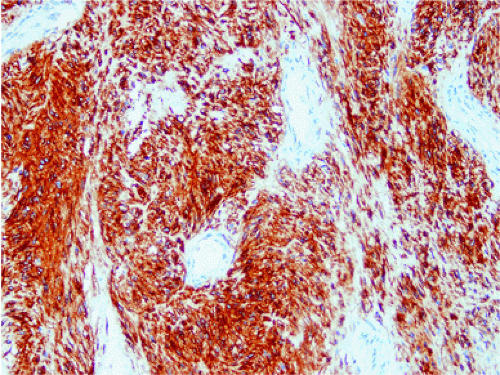 FIG. 19.25. c-kit immunostaining of a gastrointestinal stromal tumor. This tumor is strongly positive for the kit protein. |
GISTs also express the intermediate filament nestin, a protein that is not expressed in other mesenchymal tumors, unless there is neural differentiation (50). Almost 100% of GISTs strongly express the dog1 (discovered in GISTs) gene. The cell surface protein expression is highly specific for GISTs (51). PKCu is positive in up to 96% of GISTs (52). The GANT variant of GIST may show patchy immunostaining for neuron-specific enolase (NSE), S100, synaptophysin, vasoactive intestinal polypeptide, substance P, chromogranin, and neurofilament protein. Muscle cell markers are variably positive.
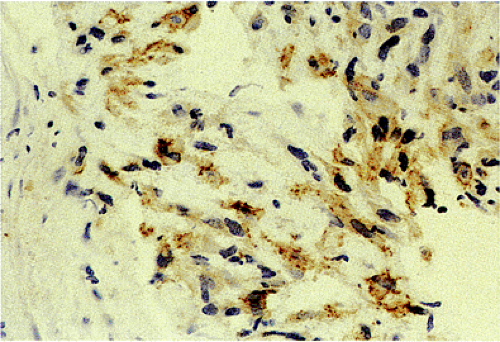 FIG. 19.26. CD34 staining of a gastric gastrointestinal stromal tumor. |
Differential Diagnosis
Because the morphologic spectrum of GISTs is wide, the differential diagnosis is also wide. It includes smooth muscle tumors, schwannomas, malignant peripheral nerve sheath tumors, solitary fibrous tumors, inflammatory fibroid tumors, synovial sarcomas, mesotheliomas, neuroendocrine tumors, glomus tumors, sarcomatoid carcinomas, malignant melanomas, and angiosarcomas. Immunohistochemical analysis is required to distinguish among the entities within this differential diagnosis (Table 19.5). CD117 expression confirms the diagnosis in most tumors. Rarely, epithelioid GISTs express CD34 in the absence of CD117. Since some epithelioid GISTs show cytokeratin immunoreactivity, coexpression of either CD34 or CD117 becomes critical to avoid misdiagnosing these cases as carcinomas. Inflammatory GISTs raise the differential diagnosis of follicular dendritic cell sarcoma, inflammatory myofibroblastic tumor, inflammatory leiomyosarcoma, inflammatory malignant fibrocytic sarcoma, and inflammatory fibroid polyps.
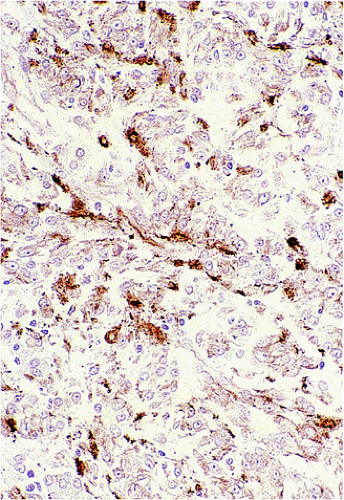 FIG. 19.27. Individual tumor cells are variably immunoreactive for actin in this gastrointestinal stromal tumor. |
TABLE 19.5 Differential Diagnosis of Gastrointestinal Stromal Tumors (GISTs) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 19.6 Gastrointestinal Stromal Tumors: Prognostic Factors | |
|---|---|
|
Prognosis
Predicting the clinical behavior of GISTs requires an evaluation of the gross, microscopic, and, possibly, immunohistochemical and molecular features of the tumors (Table 19.6). Tumor size, mitotic rate, patient age, and tumor location have been most commonly cited as the best predictors of clinical behavior at any site (26). However, some believe that tumor size and mitotic index are insufficient to provide an accurate long-term prediction of prognosis (53). Table 19.1 shows the consensus recommendations for defining risk of aggressive behavior (26), and we have previously discussed factors that appear to be important in specific anatomic locations.
Tumor size alone cannot be used to reliably separate benign from malignant gastric stromal tumors (34), since significant overlap exists between individual cases. Tumors ≥7 cm are significantly more likely to metastasize than smaller tumors. However, up to 35% of patients with an adverse outcome have tumors measuring <7 cm. Likewise, 33% of patients with a good outcome have tumors measuring ≥7 cm. Metastasis occurs in 20% of tumors <6 cm compared to 85% of those that are 6 cm or larger (44). In another study, 15% of GISTs <2.5 cm, 29% of tumors measuring 2.5 to 5 cm, 65% of tumors between 5 and 10 cm, and virtually all tumors >10 cm metastasized (54). Small intestinal tumors use a size cutoff of 5 cm (see above).
Larger tumors tend to have an especially poor prognosis, partly because surgical extirpation is difficult in such lesions and larger lesions tend to invade adjacent structures. Small size in the absence of mitotic activity does not preclude malignant behavior, however. Invasion into adjacent organs also correlates with an aggressive clinical course, as does the presence of metastases.
The impact of proliferative index is highly site dependent, being most unpredictable in the small bowel. It should be noted that there is often significant intratumoral variability in the mitotic activity. Mitotic counts should be performed in the most mitotically active areas of the tumor. Generally, mitotic counts are highest in the most cellular areas of the neoplasm. High-grade lesions (>10 mitoses/10 hpf) have the most dismal outcome, with 0% 10-year survival (55). One can measure proliferation in ways other than by mitotic counts, including immunostaining for proliferating cell nuclear antigen (PCNA) and Ki-67 as well by studying silver staining nucleolar organizing regions (AGNORs). All studies using these methods show a relationship between cell proliferation and prognosis, but they do not add anything to standard mitosis counting.
Tumor location helps predict outcome. Patients with esophageal tumors have the best survival, whereas the prognosis is worst for those with small intestinal GISTs (53). In stark contrast to gastric tumors, intestinal tumors >10 cm with mitotic activity of five or fewer mitoses per 50 hpf and those ≤5 cm with mitoses of greater than five mitoses per 50 hpf have a high metastatic rate (38). Benign GISTs outnumber malignant GISTs in the stomach, whereas malignant GISTs are more common in the intestine (38).
Tumor cell necrosis is more common in malignant than in benign GISTs, but benign tumors may contain necrotic areas, particularly if there has been torsion of pedunculated lesions. Mucosal invasion is a highly specific marker of malignancy. However, it is not a sensitive marker, since many GISTs do not infiltrate the overlying mucosa. Additionally, surface ulceration may make evaluation of this feature difficult.
Tumors in young people tend to have a better prognosis than those developing in older individuals, even when metastases are present, especially in the setting of the Carney triad. Of note, radiographic evidence of pulmonary lesions should not be taken as clinical evidence of lung metastases, especially in young women, because the pulmonary lesion may represent a benign pulmonary hamartoma in patients with the Carney triad.
The prognostic value of KIT mutations is controversial. Some have found that KIT mutations preferentially occur in malignant GISTs (56) and that the mutations have independent prognostic significance (57). Others suggest that KIT mutation is obligatory for GIST development, and therefore not related to tumor prognosis (58). Rather, the type or location of mutation may determine a patient’s prognosis and treatment response. Mutations in exon 11 associate with a poor outcome (59). Deletions in codons 557/558 of exon 11 significantly correlate with disease recurrence and shortened survival (57). Gastric GISTs with KIT exon 11 deletions have a higher rate of aggressive disease than tumors with exon 11 point mutations (11).
Treatment
Surgery is the initial treatment, with the aim to perform a complete en bloc resection of the tumor, which may include removal of adjacent organs if required. However, even after complete surgical resection, many malignant GISTs recur. When patients fail surgical treatment, they typically fail at the original tumor site, or in the peritoneum, omentum, or liver (61). These lesions may also metastasize to the ovary (62). GISTs rarely metastasize outside the abdominal cavity. Recurrences occur within the first 2 years, although low-grade tumors may not recur for decades.
Until recently, no other therapies (chemotherapy or radiation) have been effective. However, the use of targeted therapies designed to inactivate the tyrosine kinase activity of both the c-kit and PDGFRA receptors has improved patient prognosis. Imatinib, a selective adenosine triphosphate (ATP) competitive inhibitor of the tyrosine kinase activity of c-kit, decreases proliferation and induces apoptosis (63). It is very effective in treating metastatic or recurrent GISTs (64). More than 60% of patients with metastatic GISTs treated with imatinib have a major durable response, with a reduction of 70% to 90% of the tumor volume (64).
The efficacy of imatinib and the duration of the disease control may correlate with the type or location of KIT mutation. Eighty percent of tumors with exon 11 mutations are sensitive to imatinib treatment (65). Tumors with a novel point mutation in exon 14 in the kit ATP pocket also demonstrate imatinib sensitivity (66). In contrast, the KIT-D816V mutation affects the kinase catalytic domain, interfering with the imatinib-binding site, rendering the drug ineffective (67). Only 50% of tumors with exon 9 mutations respond and 20% of tumors with no mutations have a partial response. Missense mutations in the kinase domain 1 correlate with imatinib resistance (68). Cells carrying KIT-Del557-558/T670I or KIT-Ins502-503/V654A mutations are resistant to imatinib.
The location of the mutation in the PDGFRA gene also affects sensitivity to imatinib. Most isoforms with a substitution involving codon D842 in exon 18 (D842V, RD841-842KI, and D1842-8431M) are resistant to the drug, with the exception of D842Y. Other mutations in exon 18 (D846Y, N84K, Y849K, and HDSN845-848P) are imatinib sensitive (16). Corless et al recently proposed a molecular classification of GISTs (16) (Table 19.7).
It has now become clear that some patients with GISTs develop resistance to imatinib during chronic therapy. The resistance may be of two forms: Either primary resistance or acquired resistance after an initial benefit from the drug. Secondary mutations are found in 46% of tumors in patients with acquired resistance, most of whom had had a primary mutation in kit exon 11 (68). Most secondary mutations are located in exon 17; others occur in exons 13 and 14 (68,69). Progressive tumors may also show an acquired PDGFRA-D842V mutation.
The next major challenge will be the treatment of tumors that become resistant to imatinib, which will presumably occur through the selection of pre-existing drug-resistant clones that carry unresponsive mutant forms of either the kit or PDGFRA receptor. It is conceivable that a combination that uses imatinib with other agents or surgery may prove the way to prevent the development of drug-resistant tumors. New drugs that are currently being developed target the ATP binding sites of various tyrosine kinases, including c-kit, to address the insensitivity of some tumors to imatinib (70). For example, a new kinase inhibitor, SU11248, has a broader spectrum of activity, inhibiting both kit and PDGFRA as well as the vascular endothelial growth factor receptor, so that it is also a powerful antiangiogenic agent. The drug is currently in clinical trials (70).
Smooth Muscle Tumors
Leiomyomas
True gastrointestinal smooth muscle tumors exist, but they are uncommon. Leiomyomas are most common in the esophagus, anorectum, and colon. Leiomyomas constitute approximately 0.4% of all esophageal neoplasms; they are the most common benign mesenchymal esophageal tumor, followed by granular cell tumors. Esophageal leiomyomas typically affect adults; males are more commonly affected than females. Autopsy studies show very small leiomyomas (micro- or seedling leiomyomas) in approximately 8% of individuals (71). Esophageal leiomyomas develop at a younger age than GISTs, with a median age of 30 to 35 years. Esophageal leiomyomas may afflict patients with Alport syndrome or with multiple endocrine neoplasia type 1 (MEN-1) (72). We are also seeing localized leiomyomas in many of our patients undergoing resection of distal esophageal adenocarcinomas after preoperative chemoradiation. Colonic leiomyomas show a male predominance and a median age of 62.
Esophageal leiomyomas may become symptomatic or be found incidentally. Polypoid esophageal leiomyomas project into the lumen, producing retrosternal, epigastric, or noncardiac chest pain; pyrosis; and dysphagia. Esophageal tumors also cause obstruction or bleeding. Antral leiomyomas produce gastric outlet obstruction. Intestinal tumors may lead to intussusception, volvulus, and torsion. Appendiceal neoplasms remain clinically silent or produce acute appendicitis. Colonic lesions tend to remain asymptomatic, only to be discovered incidentally during the workup of the patient for occult bleeding or during colonoscopy for other reasons. Smaller colonic lesions present as polyps, especially when they originate in the muscularis mucosae (Fig. 19.28). Anorectal leiomyomas present with rectal pain, tenesmus, or constipation.
TABLE 19.7 Molecular Classification of Gastrointestinal Stromal Tumors (GISTs) | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||
Leiomyomas arise from either the muscularis mucosae or muscularis propria, where they form well-demarcated nodular muscular expansions or well-demarcated intramural tumors. They may also present as small polyps projecting into the lumen. The mucosa and serosa stretch over the surface of small lesions. Leiomyomas are typically pale, firm or rubbery, occasionally lobulated, usually round or oval pinkish-white lesions with a whorled appearance resembling their uterine counterparts. Unusual tumors measure >10 cm.
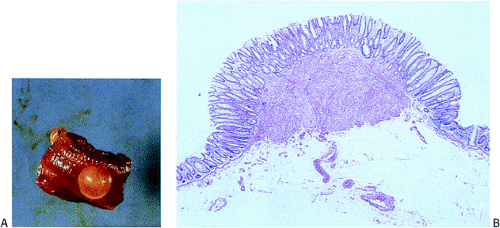 FIG. 19.28. Colonic leiomyomas. A: This colonic leiomyoma arises from the muscularis propria. B: Histologic features of a leiomyoma of the colon showing a downward proliferation of the muscularis mucosae, which elevates the mucosa. This lesion presented as a sessile polyp. |
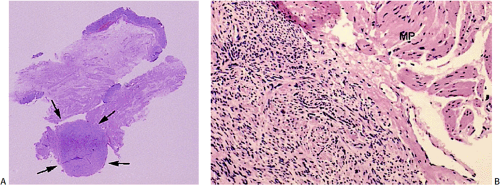 FIG. 19.29. Microleiomyoma of the stomach. A: Whole mount section of the stomach showing the presence of a gastric leiomyoma (arrows). B: Higher magnification showing the junction of the leiomyoma (lower left) and the muscularis propria (MP). |
Esophageal and gastric leiomyomas are generally small, sessile, submucosal lesions that occasionally become pedunculated. They rarely ulcerate. Most arise in the distal esophagus, with a decreasing frequency proximally. Esophageal and gastric microleiomyomas are small (<1 cm), incidentally found microscopic lesions. They appear as tiny, whitish nodules typically involving the gastroesophageal junction, where they arise from the inner oblique muscle (Fig. 19.29). However, they can be seen anywhere in the esophagus or stomach. Many colorectal leiomyomas originate from smooth muscle cells in the muscularis mucosae (Fig. 19.28). They typically measure several millimeters in diameter, although lesions up to 2.2 cm have been described (32).
Leiomyomas are low to moderately cellular tumors containing interlacing fascicles of bland spindle-shaped, smooth muscle cells. The cells are more haphazardly arranged than in the normal muscular layers; peripherally, they may merge into the adjoining muscular layer. The cells appear hypertrophic and contain an abundant eosinophilic fibrillar cytoplasm. They often display prominent nuclear palisading. Epithelioid differentiation and hyalinization may be present. The nuclei generally appear elongated and cigar shaped, usually without significant pleomorphism. Mitotic activity is minimal or absent. Perinuclear halos are often present. Vascularity varies from tumor to tumor. Duodenal leiomyomas may contain considerable nuclear atypia, but the mitotic rates are usually less than one mitosis per 50 hpf.
Colorectal tumors may contain multiple eosinophilic globules that are positive for α-smooth muscle actin, muscle-specific actin, and desmin (73). These globules differ from the skeinoid fibers seen in GISTs. The tumor cells are positive for smooth muscle actin, are variably positive for desmin, and usually fail to express c-kit and CD34. KIT mutations are absent. Small leiomyomas can be safely removed by polypectomy or local enucleation. Larger lesions may require resection.
Epstein-Barr Virus–Associated Smooth Muscle Tumors
Epstein-Barr virus (EBV)-associated smooth muscle tumors are rare lesions that develop in immunocompromised individuals, particularly pediatric AIDS patients and organ transplant recipients. The lesions typically develop in children and adults under the age of 50 (74). In transplant patients, the tumors usually develop 3 or more years following the transplant surgery. The tumors differ from conventional leiomyomas in that the tumors are frequently multiple and in many small lesions the tumors appear to arise in vascular walls.
Most gastrointestinal lesions develop in the small bowel. The tumors range in size, with many tumors being only several millimeters in diameter. The tumors are relatively well differentiated with only modest nuclear atypia. Mitotic activity ranges from virtually none to levels as high as 18/10 hpf, with an average of less than three mitoses per 10 hpf (74). Focal areas of myxoid degeneration or necrosis are present in a minority of cases. The tumors contain intratumoral T lymphocytes and foci of primitive rounded cells that arise gradually or abruptly from a background of differentiated smooth muscle cells. The presence of these primitive cells has no impact on prognosis (74). Given the multiplicity of the tumors, the question arises as to whether they represent multiple independent primary tumors (i.e., independent infection events) or metastases from a single primary tumor. Most data support multiple infection events (74).
Despite the fact that the histologic features of these tumors meet the criteria for leiomyosarcomas based on the Billings criteria (75), patients have an excellent prognosis, with few patients dying of their disease (74).
Despite the fact that the histologic features of these tumors meet the criteria for leiomyosarcomas based on the Billings criteria (75), patients have an excellent prognosis, with few patients dying of their disease (74).
Leiomyomatosis
Leiomyomatosis is the term used to describe multifocal, ill-defined, smooth muscle proliferations that resemble clusters of leiomyomas or form linear constricting masses. The disorder affects males twice as frequently as females (76). Gastrointestinal leiomyomatosis may represent an isolated disorder or it can develop in patients with several syndromes, including Alport syndrome (AS), gastroesophageal-vulvar leiomyomatosis (GVL), MEN-1 (77), tuberous sclerosis (78), tracheobronchial leiomyomatosis, and pyloric stenosis (76). GVL is a rare, indolent, multifocal smooth muscle proliferation, primarily involving the gastroesophagus and vulva; it can affect other areas as well (79). Many patients also have Alport syndrome. The multiple tumors arising in the setting of MEN-1 are independent clones, although they develop through MEN1 alterations. A hereditary defect of basement membrane is present in some patients (80).
The molecular changes of leiomyomatosis vary, depending on the background on which the tumors develop. A deletion of the COL4A5/CLL4A6 locus located on the X chromosome (81) is present in some tumors. Patients with tuberous sclerosis show mutations in either the TSC1 (chromosome 9q34) or TSC2 (chromosome 16p13) tumor suppressor gene (82).
Esophageal leiomyomatosis may cause regurgitation, vomiting, failure to thrive, cough, and stridor secondary to tracheobronchial compression. The disease progresses slowly with symptoms often being present for years before the disease is correctly diagnosed. Infiltration of the esophageal myenteric plexus by hyperplastic smooth muscle cells causes manometric features resembling achalasia. Patients with GVL may present with esophageal symptoms or intestinal pseudo-obstruction (83). Clitoral hypertrophy and vulvar and periurethral leiomyomas may also be present. One patient with tuberous sclerosis presented with multiple areas of leiomyomatosis throughout the colon and prolonged constipation (84). The intestine and bowel appeared dilated proximally and the dilation decreased distally. This patient was also mentally retarded.
Esophageal leiomyomatosis usually extends from the midesophagus to the proximal stomach. Although most of the esophagus is involved, one may see dominant masses involving only a segment of the esophagus. The muscle proliferations arise from any of the muscle layers, most often the muscularis propria. Leiomyomatosis also affects extraesophageal sites, including the tracheobronchial tree, the female genital tract, the small intestine, or the mesentery. The walls of the rectum and anal canal may be markedly thickened, and the rectum may be severely dilated.
The neoplastic proliferations present as many ill-defined, pale, sausagelike or U-shaped masses that vary in diameter. They may grow in a semi-constricting manner, sometimes affecting long gastrointestinal segments measuring up to 35 cm in length. They may also be as small as 0.5 cm in diameter. The proliferations may extend to the serosa, and may invade adjacent normal tissues. Some nodules protrude into the colonic lumen, producing sessile polyps (84).
Histologically, leiomyomatosis consists of multiple confluent or nonconfluent well-differentiated smooth muscle proliferations and nodules arising from both the muscularis mucosae and muscularis propria. The lesions extend in a pushing manner through the muscularis mucosae into the lamina propria, but do not reach the lumen. The smooth muscle proliferations often infiltrate the myenteric plexus. The cells lose their usually parallel orientation and form fascicles, interlacing bundles, and whorls. The cells contain oval, slightly enlarged hyperchromatic nuclei. Nuclear atypia and mitoses are difficult to find. Variable amounts of collagen are present. Hyperplastic neural changes sometimes accompany the smooth muscle proliferation (77). The smooth muscle cells are positive with antibodies to actin and vimentin and are variably desmin positive. They are negative with a host of other antibodies including CD34, CD117, neurofilament, glial fibrillary acidic protein, CD34, S100, synaptophysin, CK/AE1-3, NSE, chromogranin, vasoactive intestinal polypeptide, CD31, estrogen receptor, and progesterone receptor.
Leiomyomatosis differs from multiple leiomyomas in that multiple leiomyomas are multiple circumscribed tumors with distinct pushing margins rather than poorly delineated smooth muscle proliferations. Leiomyomatosis also differs from idiopathic muscular hypertrophy, which is a rare condition in which the entire muscularis propria becomes uniformly thickened without nodule formation. GVL behaves in an indolent manner and is unlikely to be malignant. Patients with extensive leiomyomatosis may benefit from surgical resection and esophageal replacement.
Disseminated Peritoneal Leiomyomatosis (Leiomyomatosis Peritonealis Disseminata)
Disseminated peritoneal leiomyomatosis (DPL) primarily affects women in the reproductive age group, many of whom are pregnant, postpartum, or taking oral contraceptives (85). Symptoms (pelvic pain and abnormal uterine bleeding) usually result from coexisting uterine leiomyomata and not from the GI lesions. Innumerable small (several millimeters in size) smooth muscle proliferations ranging from 0.1 to 2 cm stud the serosal and peritoneal surfaces and the omentum (Fig. 19.30). There may also be larger leiomyomatous masses. These nodules consist of interwoven fascicles of smooth muscle cells that exist alone or coexist with areas of endometrial or endocervical differentiation (85,86). The proliferating cells interdigitate with one another and with proliferating fibroblasts and myofibroblasts. The lesions may show mild to moderate hypercellularity, but without coagulative necrosis, pleomorphism, or cytologic or nuclear
atypia. Mitotic figures are rare (less than two mitoses per 100 hpf). Atypical mitoses are absent. The histogenesis of DPL remains controversial, but most accept that it represents a hormonally driven metaplasia of the subcelomic mesenchyme (85). Increased levels of estrogen and progesterone receptors are seen in the smooth muscle cells (85). Most DPL cases behave in a clinically benign fashion and, in some cases, the lesions may partially or completely regress (85). Occasionally, however, DPL may progress, recur, or undergo malignant transformation (87).
atypia. Mitotic figures are rare (less than two mitoses per 100 hpf). Atypical mitoses are absent. The histogenesis of DPL remains controversial, but most accept that it represents a hormonally driven metaplasia of the subcelomic mesenchyme (85). Increased levels of estrogen and progesterone receptors are seen in the smooth muscle cells (85). Most DPL cases behave in a clinically benign fashion and, in some cases, the lesions may partially or completely regress (85). Occasionally, however, DPL may progress, recur, or undergo malignant transformation (87).
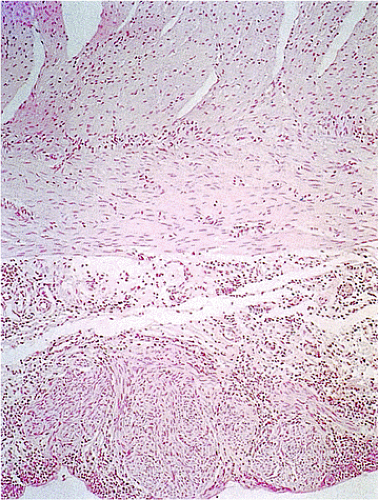 FIG. 19.30. Leiomyomatosis peritonealis disseminata. Spindle-shaped proliferating smooth muscle cells are situated between the muscularis propria and the serosa. |
Smooth Muscle Hamartomas
Smooth muscle hamartomas develop spontaneously or in the setting of tuberous sclerosis or Cowden syndrome. They present as single or multiple nodular, sessile, or, more rarely, pedunculated intestinal polyps. The hamartomas consist of proliferating, mature, spindle-shaped smooth muscle cells arranged in interlacing bundles that replace the lamina propria and entrap the epithelium (Fig. 19.31). There may also be fibrous tissue proliferation.
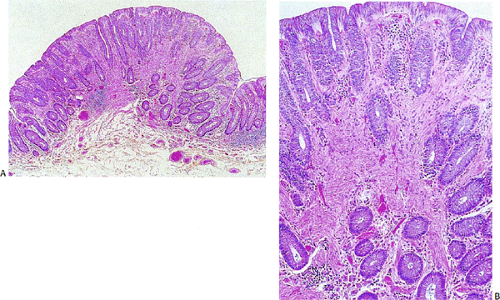 FIG. 19.31. Colonic smooth muscle hamartoma. A: Hamartomatous smooth muscle proliferation involving colonic lamina propria. B: Higher-power view demonstrating admixed muscular and fibrous tissue displacing the colonic glands. Note the lack of cellular atypia. |
Smooth muscle hamartomas differ from Peutz-Jeghers–associated hamartomas in that the latter contain prominent arborizing smooth muscle proliferations that divide the epithelium and its surrounding normal-appearing lamina propria into segments. In contrast, smooth muscle hamartomas show proliferating smooth muscle cells obliterating
the lamina propria lying between the crypts without the prominent smooth muscle arborizations. Hamartomas also differ from leiomyomas in that leiomyomas are generally solitary and well circumscribed.
the lamina propria lying between the crypts without the prominent smooth muscle arborizations. Hamartomas also differ from leiomyomas in that leiomyomas are generally solitary and well circumscribed.
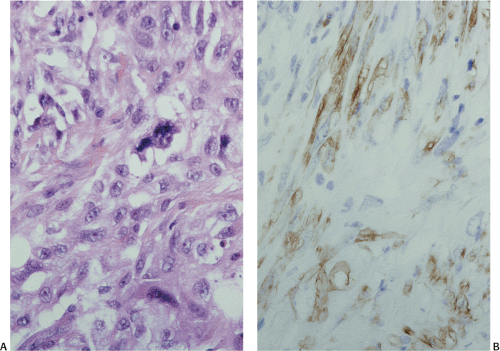 FIG. 19.32. Small intestinal leiomyosarcoma. A: These spindle cell lesions typically show much more pleomorphism and atypical mitoses than malignant gastrointestinal stromal tumors. B: Actin immunostain of the same lesion. The tumor was kit negative. |
Leiomyosarcomas
True leiomyosarcomas do develop in the luminal gut, the omentum, and the mesentery, but they are not nearly as common as GISTs. Leiomyosarcomas, particularly vascular ones, affect older individuals. Leiomyosarcomas generally present in the same way as malignant GISTs, often as luminal, sometimes ulcerated, polypoid tumors (40). Grossly, the lesions appear lobular, gray-beige with pinkish tan areas and greenish areas of necrosis (50). The tumors range in size from 10 to 23 cm and may be multiple.
Gastrointestinal, omental, and mesenteric leiomyosarcomas contain well-differentiated smooth muscle cells with elongated or oval, often blunt-ended (cigar-shaped) atypical nuclei and eosinophilic cytoplasm. The tumors resemble soft tissue leiomyosarcomas. All of the tumors exhibit nuclear pleomorphism, which, unlike GISTs, can be extensive (Fig. 19.32). Coagulative necrosis is usually present; skeinoid fibers are absent. Mitotic activity is often high (sometimes >50 mitoses/50 hpf) (88). The differential diagnosis includes the entities listed in Table 19.5. Leiomyosarcomas are globally positive for α-smooth muscle actin and variably positive for desmin. They may be focally positive for cytokeratin-18, but negative for cytokeratin-19. The tumors are negative for CD34, CD117, S110, glial fibrillary acidic protein (GFAP), and other neural markers. Leiomyosarcomas lack KIT mutations.
Leiomyosarcomas are treated with surgical resection. Lesions that are completely excised may be cured. However, tumors with positive tumor margins tend to recur locally as well as metastasize to the liver, causing patient death (88).
Neural Tumors
Primary gastrointestinal neurogenic tumors are rare. They fall into two major groups: Those of peripheral nerve sheath origin (schwannomas, neurofibromas, ganglioneuromas, neuromas, and perineuromas) and those arising from the sympathetic or chromaffin system (neuroblastomas, ganglioneuromas, and paragangliomas).
Schwann Cell Tumors
Isolated gastrointestinal schwannomas constitute only 2% to 6% of all GI mesenchymal tumors and 0.2% of all gastric tumors. Patients range in age from 10 to 81 years with an average age of 52.6 (89). Women are slightly more commonly affected than men. Gastrointestinal Schwann cell
tumors are particularly common in NF1 patients (90). Some patients have associated lesions (conditions), including colonic adenomas or Gorlin syndrome (90). The clinical features vary depending on lesional size and location. They present in the same ways as GISTs. Schwannomas preferentially affect the stomach (91) followed by the colon and rectum (92). Less commonly, they arise in the small intestine and esophagus. A subset of schwannomas, the benign mucosal epithelioid nerve sheath tumors, preferentially affects the colon (93), although this may reflect the large number of colonic biopsies that are examined annually.
tumors are particularly common in NF1 patients (90). Some patients have associated lesions (conditions), including colonic adenomas or Gorlin syndrome (90). The clinical features vary depending on lesional size and location. They present in the same ways as GISTs. Schwannomas preferentially affect the stomach (91) followed by the colon and rectum (92). Less commonly, they arise in the small intestine and esophagus. A subset of schwannomas, the benign mucosal epithelioid nerve sheath tumors, preferentially affects the colon (93), although this may reflect the large number of colonic biopsies that are examined annually.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


