Management of Transplantation Renal Bone Disease: Interplay of Bone Mineral Density and Decisions Regarding Bisphosphonate Use
Paul D. Miller*
Elizabeth Shane†
*Department of Medicine, University of Colorado Health Science Center, Denver, Colorado 80262; and Colorado Center for Bone Research, Lakewood, Colorado, 80227;
†Department of Medicine, Columbia University College of Physicians and Surgeons, New York, New York 10032
INTRODUCTION
The intention of this chapter, “Management of Transplantation Renal Bone Disease,” is both to review evidence-based data on what is known about the skeletal processes that take place before and after renal transplantation, and to provide the opinion(s) of the authors regarding practical clinical management that hopefully will help guide the nephrologist in caring for these patients. We acknowledge that scientific data that permit one to extrapolate to clinical practice are limited and, in that regard, our chapter is both a challenge to write as well as a call to conduct the clinical research studies necessary to develop better evidence-based guidelines.
EPIDEMIOLOGY
The number of renal transplantations performed each year, as well as the survival of both the graft and the transplant recipient, are increasing (1, 2, 3). Bone mineral density (BMD), as measured by dual energy x-ray absorptiometry (DXA), declines in the vast majority of patients who receive no therapy to prevent bone loss (4, 5, 6, 7, 8). Fractures of the vertebrae, hip and other skeletal sites also occur, many during the first year after transplantation, as well as the later posttransplant period. In addition, nonvertebral fractures may be relatively more common in the renal transplant recipient than in other types of organ transplantation (9, 10, 11, 12, 13, 14, 15, 16, 17, 18).
NATURAL BIOLOGY OF POSTTRANSPLANTATION RENAL BONE DISEASE: INFLUENCE OF PRETRANSPLANTATION HISTOMORPHOMETRY ON POSTTRANSPLANTATION BONE LOSS
Renal bone disease (“renal osteodystrophy”) is a heterogeneous group of metabolic bone diseases with distinct clinical, pathophysiologic and quantitative histomorphometric features (Table 23.1) (19, 20, 21, 22). The two most common forms are hyperparathyroidism and adynamic bone disease. Published studies on the natural history of postrenal transplantation bone disease have generally not distinguished among groups of patients based upon their pretransplantation bone disease.
However, it is certainly possible that patients who come to kidney transplantation with severe hyperparathyroidism may differ from those with adynamic bone disease with respect to posttransplantation bone loss and fracture rates. Although biochemical tests, particularly measurement of serum parathyroid hormone (PTH) and bone specific alkaline phosphatase (BSAP), can distinguish among different forms of renal osteodystrophy in groups of patients, individual patients may be misdiagnosed if classification is based solely on biochemical parameters (23, 24). Transiliac crest bone biopsy after double tetracycline labeling with quantitative bone histomorphometry is the only reliable way to establish the type of renal bone disease in individual patients (25, 26, 27).
However, it is certainly possible that patients who come to kidney transplantation with severe hyperparathyroidism may differ from those with adynamic bone disease with respect to posttransplantation bone loss and fracture rates. Although biochemical tests, particularly measurement of serum parathyroid hormone (PTH) and bone specific alkaline phosphatase (BSAP), can distinguish among different forms of renal osteodystrophy in groups of patients, individual patients may be misdiagnosed if classification is based solely on biochemical parameters (23, 24). Transiliac crest bone biopsy after double tetracycline labeling with quantitative bone histomorphometry is the only reliable way to establish the type of renal bone disease in individual patients (25, 26, 27).
TABLE 23.1. Metabolic bone diseases seen in renal failure | |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||
In general, markedly elevated serum PTH levels (greater than six times the upper limit of normal) are associated with histomorphometric evidence of hyperparathyroidism, such as osteitis fibrosa cystica. However, there are well-known difficulties in the interpretation of PTH levels in the setting of renal disease. PTH levels may vary widely in the same individual, related to the serum calcium and/or phosphate concentrations at the time of sampling (28) (Fig. 23.1). Serum calcium and/or phosphorus are greatly influenced by the calcium concentration in the dialysis bath, oral calcium intake, vitamin D status or replacement with 1,25(OH)2 vitamin D {1,25(OH)2D}, and in the case of serum phosphate, use of binders to reduce intestinal absorption. Therefore, a single measurement of PTH may be misleading in the individual patient (27, 28, 29, 30).
 FIG. 23.1 The influence of the serum phosphorus or calcium on parathyroid hormone values. (From Llach F, Yudd M. The importance of hyperphosphatemia in the severity of hyperparathyroidism and its treatment in patients with chronic renal failure. Nephrol Dial Transplantation1998;13[Suppl 3]:57-61, with permission.) |
Another difficulty relates to the specific PTH assay used. It has long been known that patients with chronic renal failure retain carboxyterminal fragments of PTH. Recent data suggest that some of these fragments, such as the 7-84 amino acid fragment, have biologic activity that opposes the intact 1-84 molecule. Moreover, the older IRMA “intact” PTH assay overestimates the amount of bioactive “whole” PTH(1-84) in the circulation, because it also detects carboxyterminal fragments, including the 7-84 amino acid fragment. The amount of 7-84 fragment can be estimated by simultaneous measurement of both the 1-84 “whole” PTH as well as the traditional “intact” PTH assay, subtracting the “whole” from the “intact” PTH. The ratio of the two PTH forms (1-84/7-84) has been used to predict bone turnover in patients with end-stage renal disease (ESRD). A ratio of <1.0 has been associated with a high probability of adynamic bone disease (31) (Fig. 23.2), while >1.0 predicts normal or high bone turnover. If substantiated by future studies, measuring both PTH assays may improve the assessment of bone turnover and provide a noninvasive means of distinguishing between hyperparathyroid and adynamic bone disease.
Adynamic renal bone disease, a form of very low turnover bone disease with increasing prevalence in ESRD patients, is associated with an increased risk of fracture (31, 32, 33, 34, 35, 36). Adynamic bone disease requires special consideration in the ESRD and posttransplantation patient populations, since there is widespread concern among nephrologists that further suppression of bone turnover by bisphosphonates in patients with preexisting adynamic renal bone disease might adversely affect repair of microfractures and increase fracture risk (37, 38). While this is as yet a theoretical concern in humans, there are animal data suggesting that oversuppression of bone turnover by bisphosphonates may be associated with impaired microfracture repair (39, 40), negatively affecting bone strength. However, recent data in humans (41) and animals (42) suggest that, even with substantial suppression of bone turnover, strength may not be compromised. Unfortunately, there are few data on the evolution of posttransplantation renal bone loss or the response to pharmacological therapy to prevent such bone loss as a function of the type of renal osteodystrophy present before transplantation. However, we are concerned that withholding bisphosphonate therapy from renal transplant recipients, particularly during the first posttransplant year, because of fear of exacerbating adynamic bone disease, may place such patients at unnecessary risk for rapid bone loss and fracture. Studies that address this issue are urgently needed.
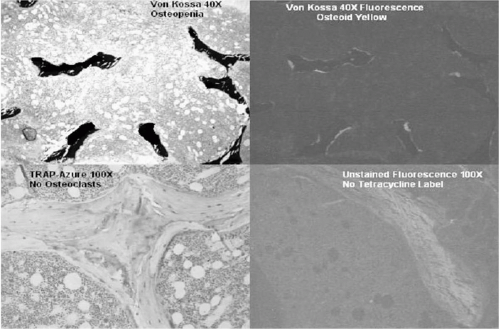 FIG. 23.2 The features of adynamic bone disease: no tetracycline uptake, little osteoid, low trabecular bone volume, and few to absent osteoclasts (Courtesy of P. Miller). |
Most forms of renal osteodystrophy may improve after transplantation (hyperparathyroidism, aluminum bone disease, and those associated with chronic metabolic acidosis, heparin exposure, and 1,25(OH)2D deficiency). In contrast, there is no evidence that adynamic bone disease improves after transplantation, and recent data suggest deterioration in histomorphometric parameters that assess bone formation, such as osteoblast surface and number, adjusted apposition rate and mineralization lag time (34,37). The lack of improvement in pretransplant adynamic bone disease after transplantation has been attributed to glucocorticoids, agents that have well-known inhibitory effects on bone formation (34,37). In one paired biopsy study (43), (before and after transplantation, without pharmacological intervention), posttransplant osteoblast surface correlated negatively with glucocorticoid dose and positively with serum PTH, while serum phosphate correlated positively with osteoblast number and positively with apoptotic osteoblasts. Paired bone biopsy studies with larger sample sizes are needed to clarify the influence of the type of pretransplant renal bone disease and subsequent therapy(s) on posttransplantation bone histomorphometry.
BONE LOSS AFTER TRANSPLANTATION: NATURAL HISTORY AND MECHANISMS
While there are limited data on the natural biology of posttransplantation bone disease as a function of histomorphometrically defined pretransplantation renal osteodystrophy, there are considerable data available on rates of bone loss and fracture after transplantation. First, it should be recognized that the prevalence of low BMD at the spine, hip, and distal radius is high even before transplantation. Prevalent vertebral fractures have been reported in 21% of the patients with ESRD, and the risk of hip fractures in this population is increased by 34%. This risk is much greater than published fracture prevalence rates in untreated postmenopausal women or elderly men without renal failure. After transplantation, most of the bone loss occurs in the first months of the first year, and the fracture incidence is also high during this period (Fig. 23.3) (7, 8, 9,17,18,44, 45, 46, 47, 48, 49, 50, 51). Fracture risk after transplantation is greater in older patients, women, diabetics, whites, and in those with lower PTH levels.
The early loss of BMD and early fracture rates after transplantation strongly suggests the greatest impact on the skeleton after transplantation is due to the adverse effects of glucocorticoids (Fig. 23.4). This hypothesis is also
supported by paired biopsy studies, cited above (43). Glucocorticoid doses are higher in the immediate posttransplant period and during rejection episodes. The effects of glucocorticoids on bone metabolism are dose-related, though even “low-dose” glucocorticoid regimens (2.5-7.5 mg prednisone/day) are associated with increased risk of fracture (Fig. 23.5) (52, 53, 54). Glucocorticoids have profound inhibitory effects on bone remodeling, with far greater negative effects on osteoblast and osteocyte life span and function than on osteoclast activity (55, 56, 57, 58). Osteoclast activity is, however, also increased by glucocorticoid administration because of its effect on the osteoprotegerin (OPG)/Receptor Activated Nuclear K-ligand (RANKL) system. RANKL promotes osteoclastogenesis by binding to its receptor RANK on osteoclasts. (Fig. 23.6) (59). OPG is a decoy receptor that binds to RANKL, preventing RANKL from binding to RANK. Glucocorticoids increase expression of RANK and decrease expression of OPG.
supported by paired biopsy studies, cited above (43). Glucocorticoid doses are higher in the immediate posttransplant period and during rejection episodes. The effects of glucocorticoids on bone metabolism are dose-related, though even “low-dose” glucocorticoid regimens (2.5-7.5 mg prednisone/day) are associated with increased risk of fracture (Fig. 23.5) (52, 53, 54). Glucocorticoids have profound inhibitory effects on bone remodeling, with far greater negative effects on osteoblast and osteocyte life span and function than on osteoclast activity (55, 56, 57, 58). Osteoclast activity is, however, also increased by glucocorticoid administration because of its effect on the osteoprotegerin (OPG)/Receptor Activated Nuclear K-ligand (RANKL) system. RANKL promotes osteoclastogenesis by binding to its receptor RANK on osteoclasts. (Fig. 23.6) (59). OPG is a decoy receptor that binds to RANKL, preventing RANKL from binding to RANK. Glucocorticoids increase expression of RANK and decrease expression of OPG.
 FIG. 23.3 Bone loss occurs rapidly after transplantation. |
 FIG. 23.4 Mechanisms of glucocorticoid-induced bone loss. |
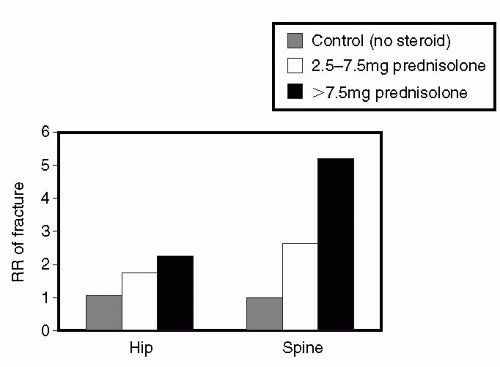 FIG. 23.5 Fractures due to glucocorticoids are greater the higher the dose but still occur at “low doses.” (From Ref. 55, with permission.) |
 FIG. 23.6 The RANK/RANK-ligand system: Glucocorticoids increase RANK-ligand production which then induces osteoclastogenesis. (From Ref. 59, with permission.) |
The effect of glucocorticoids on bone mass and fracture susceptibility may be at least partially reversible after discontinuation. Thus, many transplant programs attempt to discontinue glucocorticoids after several years of stable renal function, while maintaining graft survival with other immunosuppressive agents (60). However, it is not clear whether discontinuing glucocorticoids also means that treatment for glucocorticoid-induced bone loss (such as bisphosphonate therapy) can also be discontinued. This important consideration will be discussed in a later section of this chapter.
Cyclosporin A (CsA) and tacrolimus (FK506) are both calcineurin inhibitors. Calcineurin inhibitors inhibit T-cell function and have direct effects on calcineurin genes expressed in osteoclasts (61, 62). In the rat, CsA leads to rapid bone loss associated with increased bone turnover. It is difficult to elucidate the effects of CsA in humans, as they are most often given together with glucocorticoids, although similar effects of CsA alone have also been described in humans after renal transplantation (63,64). Although there is contrasting evidence that in a steroid-free environment, CsA may not have negative effects on bone (65, 66, 67), the transiliac bone biopsy sections shown in Figures 23.7 and 23.8 are from a woman 15 years after renal transplantation with a stable serum creatinine of 1.3 mg/dL and multiple fractures. This woman never received glucocorticoids. She has biochemical evidence of very high bone turnover, despite normal PTH and PTHrP levels. A biochemical marker of bone resorption, urinary excretion of N-telopeptide of collagen cross-links (N-telopeptide, NTX) was markedly and consistently elevated (400-600 nmol BCE/mmol creatinine; normal <70). Her serum BSAP, a marker of bone formation and turnover, was also markedly elevated (90-100 ng/mL;
normal <24). The biochemical evidence of increased bone turnover is corroborated by the biopsy results. Patients such as this woman certainly suggest that CsA alone may induce high bone turnover and clinically severe bone disease in renal transplant recipients. With regard to treatment, animal studies demonstrate that antiresorptive drugs reduce the high bone turnover induced by CsA (68, 69, 70, 71, 72).
normal <24). The biochemical evidence of increased bone turnover is corroborated by the biopsy results. Patients such as this woman certainly suggest that CsA alone may induce high bone turnover and clinically severe bone disease in renal transplant recipients. With regard to treatment, animal studies demonstrate that antiresorptive drugs reduce the high bone turnover induced by CsA (68, 69, 70, 71, 72).
 FIGS. 23.7 and 23.8 Bone histomorphometry of a woman 15 years posttransplant who has never received glucocorticoids but 15 years of cyclosporin: features of excessive high bone turnover. (Courtesy of Paul Miller and William Huffer.) |
FK506 also causes trabecular bone loss in the rat (73, 74, 75, 76), although perhaps less severe than CsA (72). Fewer studies have evaluated the skeletal effects of FK506 in humans,
although rapid bone loss has been reported (73,74) with FK506-based immunosuppression. However, FK506 may also cause less bone loss in humans than CsA (75,76), perhaps by permitting the use of lower glucocorticoid doses. There are even fewer data regarding the skeletal effects of the other immunosuppressive agents such as azathioprine, sirolimus (rapamycin), mycophenolate mofetil, and daclizumab (77, 78, 79).
although rapid bone loss has been reported (73,74) with FK506-based immunosuppression. However, FK506 may also cause less bone loss in humans than CsA (75,76), perhaps by permitting the use of lower glucocorticoid doses. There are even fewer data regarding the skeletal effects of the other immunosuppressive agents such as azathioprine, sirolimus (rapamycin), mycophenolate mofetil, and daclizumab (77, 78, 79).
Bone disease after kidney transplantation may also be related to factors other than glucocorticoids and calcineurin inhibitors. Among these factors are persistent hyperparathyroidism, osteomalacia, hypophosphatemia, metabolic acidosis, especially forms of renal tubular acidosis (80, 81, 82, 83, 84). These conditions may cause bone loss and fracture(s) that may be attributed incorrectly to immunosuppressive drugs, yet are often separate and correctable. In the immediate transplantation period with a well functioning graft, hyperparathyroidism, hypophosphatemia, and low serum 1,25(OH)2D resolve in some, though not in all patients (85). Persistent hyperparathyroidism, and the hypercalcemia that may accompany it, may jeopardize the grafted kidney, as there exists a linear relationship between the height of serum calcium and reduction in glomerular filtration rate (GFR) (59). In some patients with very severe pretransplant hyperparathyroidism, there may be inconsistent resolution of parathyroid hyperplasia after transplantation, despite increased endogenous production of 1,25(OH)2D and resolution of hyperphosphatemia, that may contribute to their loss of bone mass (86). Guidelines have been developed for performing parathyroidectomy (PTX) in patients with asymptomatic and symptomatic primary hyperparathyroidism (87, 88, 89). It is less clear when to perform PTX in renal transplant recipients, in whom justification for PTX is based upon the degree of hypercalcemia, since the primary goal in this population is preservation of GFR. The American Society of Transplant Clinical Practice Guidelines recommend PTX for serum calcium levels persistently >11.5 mg/dL (60). However, other considerations may also be important, such as rates of ongoing bone loss, recently shown to be greater in renal transplant recipients with high serum PTH levels and biochemical evidence of increased bone turnover (90).
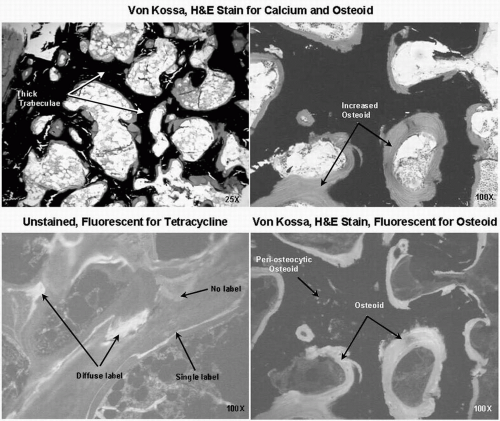 FIG. 23.9 Osteomalacia: features of abundant and thick osteoid. (Courtesy of Paul Miller and William Huffer.) |
Low serum levels of either 25-OHD or 1,25(OH)2D can also persist after transplantation (90,91). Vitamin D deficiency (serum 25-OHD <10 ng/mL) or insufficiency (<30 ng/mL) may lead not only to calcium malabsorption and secondary hyperparathyroidism, but also to mineralization defects that may result in frank osteomalacia (Fig. 23.9) (83, 84, 85,90, 91, 92). An elevated BSAP level is not specific for the type of renal bone disease and could suggest hyperparathyroidism, frank histological osteomalacia, or another high bone turnover state without osteomalacia. Persistent elevation
of BSAP after renal transplantation requires an evaluation to determine the etiology, which may require bone biopsy with quantitative bone histomorphometry to make the correct diagnosis.
of BSAP after renal transplantation requires an evaluation to determine the etiology, which may require bone biopsy with quantitative bone histomorphometry to make the correct diagnosis.
Skeletal metabolism may also be affected by the development of hypophosphatemia after renal transplantation (93,94). Hypophosphatemia may be related to persistent hyperparathyroidism in some cases and may improve gradually with resolution of the hyperparathyroid state. However, hypophosphatemia may also occur in the absence of hyperparathyroidism, perhaps due to intestinal malabsorption, a renal phosphate leak in the transplanted kidney, or to a primary defect in phosphate handling, related to one or more of the recently described factors that control phosphate homeostasis (phosphatonins).
Related posts:
 Detection of Recipient Pretransplant Alloreactivity
Detection of Recipient Pretransplant Alloreactivity
 Surgical Issues in the Transplant Recipient
Psychiatric and Psychosocial Issues in Kidney Transplantation
Surgical Issues in the Transplant Recipient
Psychiatric and Psychosocial Issues in Kidney Transplantation
 Hematologic Complications of Transplantation
Hematologic Complications of Transplantation
 Cytomegalovirus in Renal Transplantation
Cytomegalovirus in Renal Transplantation
 Medical Complications of the Eyes, Nasopharynx, Dentition, Oropharynx, and Hearing in the Kidney Transplant Recipient
Medical Complications of the Eyes, Nasopharynx, Dentition, Oropharynx, and Hearing in the Kidney Transplant Recipient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


