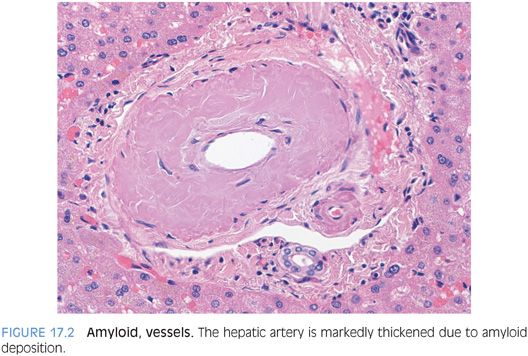
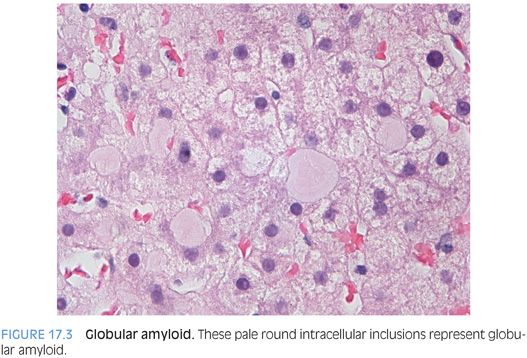
The specific type of amyloid cannot be determined by the hematoxylin and eosin (H&E) findings. However, amyloid deposits can be further subtyped on liver biopsy by immunostains, by laser microdissection and mass spectrometry on formalin-fixed, paraffin-embedded tissues,2 or by laboratory testing of other body tissues. Most cases of amyloidosis in the liver are associated with plasma cell dyscrasias, but amyloid deposits can be associated with a wide range of inflammatory and inherited conditions.
Stains and Other Ancillary Studies
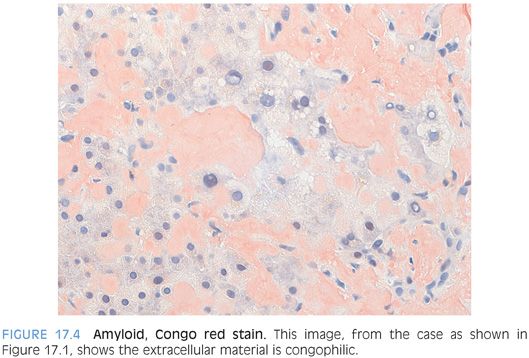
A Congo red stain is necessary to confirm the diagnosis of amyloid disease. By routine light microscopy, the amyloid deposits should demonstrate a distinctive red-orange color termed congophilia (Fig. 17.4). Polarization will then demonstrate “apple-green” birefringence (eFig. 17.5). You may have to polarize in a dark room to optimally see the birefringence. Even in the best of cases, the apple-green color tends to be somewhat pale and patchy. As an important diagnostic pitfall, the normal collagen in portal tracts will also polarize. However, the lack of congophilia will allow separation from true amyloid. In addition, the quality of the birefringence color on polarization of collagen fibers tends to be different, with a silvery white color that lacks the yellow-green color of true amyloid.
Additional immunostains to determine the type of amyloid disease can also be performed. In some centers, these stains are an integral part of the workup for amyloid disease, whereas in other centers, the amyloid is worked up mostly or entirely from the clinical side. Both approaches work fine.
AMYLOID LIKE MATERIAL THAT IS CONGO RED NEGATIVE. In this situation, the first step is to repeat the Congo red stain and make sure the slide was cut to the appropriate thickness (typically 10 μm instead of the usual 4 μm for light microscopy). If the stain is still negative, then consider the possibility of Waldenström macroglobulinemia, which is associated with deposits of immunoglobulin M (IgM) heavy chains. The deposits in Waldenström macroglobulinemia can closely resemble amyloid but are Congo red–negative.3 Deposits are kappa light chain–restricted (if they are lambda light chain–restricted, classical amyloid disease is more likely).
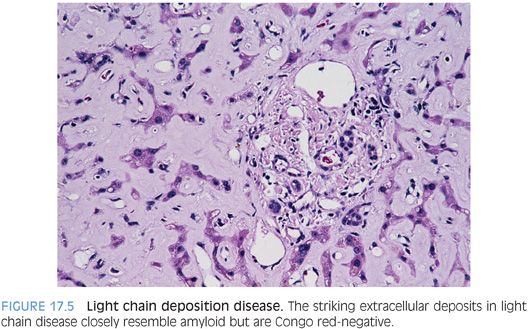
Light chain deposition disease is a second diagnosis to consider when there are extracellular deposits that resemble amyloid (Fig. 17.5) but are Congo red–negative. Light chain deposition disease is typically associated with renal disease, but sometimes, liver disease can be the first clinical manifestation. The deposits tend to be diffuse and heavy with a strong sinusoidal pattern but are Congo red–negative.4 They still have a beta-sheet pattern at the ultrastructural level, but like the deposits in Waldenström macroglobulinemia, they lack the congophilic staining and birefringence on Congo red stain.
Currently, there is some debate on whether the term amyloid should be used only for cases that are Congo red–positive or whether the term can be reasonably extended to any extracellular proteinaceous deposits that have polymerized to form beta-pleated sheets on ultrastructure, even if they are Congo red–negative.
SYSTEMIC AUTOIMMUNE CONDITIONS
Mild increases in liver enzymes are common in many systemic autoimmune conditions, including common variable immunodeficiency, systemic lupus erythematosus, rheumatoid arthritis, and juvenile rheumatoid arthritis. The enzyme elevations can be persistent or intermittent and sometimes prompt liver biopsies. The liver biopsies most commonly show a mild nonspecific lymphocytic hepatitis involving the portal tracts with minimal or mild lobular lymphocytic inflammation. There is no evidence that the mild inflammatory changes in these situations lead to liver fibrosis or cirrhosis. If there are more than mild inflammatory changes, biliary tract changes, fatty liver, cholestasis, or clear fibrosis, then a superimposed or coexisting liver disease is likely. True autoimmune hepatitis can also be seen in these conditions, but it will look histologically like a typical autoimmune hepatitis and will be accompanied by positive serologies for antinuclear antibody (ANA) and/or antismooth muscle antibodies as well as elevated serum immunoglobulin G (IgG) levels. Similarly, chronic biliary tract diseases may also co-occur by chance and should be diagnosed using the usual features. A drug effect can be particularly hard to exclude with a mild nonspecific hepatitis because many times patients will be taking numerous medications. Sometimes, a temporal correlation can be established with liver enzyme elevations and a recently added drug.
CYSTIC FIBROSIS
Cystic fibrosis is the most common inherited disease in Caucasians, with a frequency of 1 in 3,000 live births. The disease is caused by mutations in the CFTR gene, with the most common mutation being ΔF508. The CFTR protein is a glycoprotein located at the apical end of secretory cells that allows chloride transport from the cells into the lumens of various anatomic structures. Mutations lead to thick viscous secretions in the airways, intestine, pancreas, and biliary tree. In some cases, the diagnosis is first suggested by the liver pathology findings.5
Elevated levels of aspartate aminotransferase (AST), alanine aminotransferase (ALT), and γ-glutamyltransferase (GGT) are common and generally mild, with levels typically less than 2.5 times the upper limit of normal. Transient enzyme elevations are common in the first 3 months of life (approximately 50% of cases) but typically resolve.6 However, persistent enzyme elevations often develop in those aged 10 years and older, affecting approximately 40% of older children and young adults.6 Hepatomegaly, elevated liver enzymes, splenomegaly, and esophageal varices are the most common clinical indications for liver biopsy.7
Liver biopsies reveal pathology in the majority of cases (approximately 80%), although pathology is more likely to be present in cases with abnormal liver enzymes.6,8 The portal tracts can show a pattern of mild chronic biliary tract obstruction, with patchy bile ductular proliferation and nonspecific portal chronic inflammation. In some cases, especially with more advanced fibrosis, the ductular proliferation can be brisk and be accompanied by neutrophilic inflammation.6 Despite the history of cystic fibrosis, inspissated secretions in the bile ducts are rarely seen, present in approximately 5% of cases overall,7 and are patchy when present.
The portal tracts can also show loss of the portal veins in some cases. In these cases, the liver parenchyma can demonstrate changes of nodular regenerative hyperplasia. Nodular regenerative hyperplasia has been reported by only a few groups but may be an underappreciated aspect of the pathology of cystic fibrosis.9,10 Nodular regenerative hyperplasia can be associated with portal hypertension despite the lack of significant fibrosis.
The most common finding in the hepatic lobules is macrovesicular steatosis. At least some degree of fatty change is seen in approximately 65% of cases, with moderate or severe fatty change in 25% to 35% of cases.6,7 The etiology of the fatty change is unclear and its role, if any, in fibrosis progression is also unknown. In most cases, the fibrosis appears driven by biliary pathology and not the steatosis. In keeping with this, steatohepatitis is uncommon.
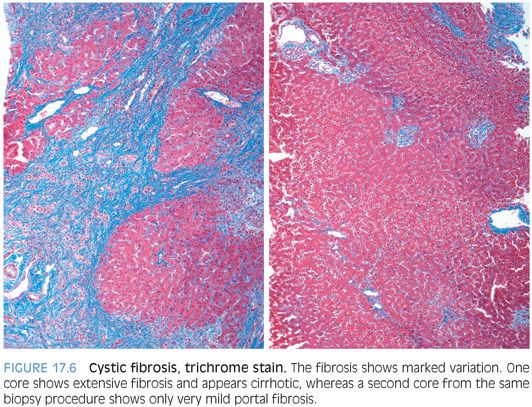
Early fibrosis is portal-based and often seen in association with portal tracts that show mixed lymphocytic and neutrophilic inflammation and a bile ductular proliferation. The fibrosis can be marked in one part of the biopsy and minimal or absent in others, a pattern that is called focal biliary cirrhosis in the cystic fibrosis literature (Fig. 17.6). The fibrosis in other cases can be advanced, with bridging fibrosis or established cirrhosis. These more severe stages of fibrosis can still retain an element of patchiness, with areas of the biopsy that appear relatively normal. Fibrosis can rarely be severe in infancy, especially in children with meconium ileus, but in most cases, fibrosis begins in late childhood and teenage years.6 Moderate or severe fibrosis is seen approximately 40% to 60% of individuals with persistently abnormal liver enzymes, representing 10% of all individuals with cystic fibrosis.6,7 Of note, advanced fibrosis can be seen even if liver enzymes are normal or near normal.11 Fibrosis on liver biopsy predicts portal hypertension.12
Other liver-related findings include biliary strictures, both intrahepatic and extrahepatic. By magnetic resonance cholangiopancreatography, biliary pathology is seen in almost all patients with clinical liver disease and one-half of those without clinical liver disease. The imaging findings include bile duct dilatation, focal strictures, narrowed areas, and beading, with alternating areas of stricture and dilatation.13 The extrahepatic strictures often involve the common hepatic duct as a result of pancreatic disease. A microgallbladder is present in about one-fourth of cases, and cholelithiasis develops in 10%.
DIABETES MELLITUS
There are three main patterns of liver injury seen in liver biopsies of patients with diabetes and abnormal liver enzymes: Glycogenic hepatopathy and fatty change are the most common, whereas diabetic sclerosis is rare.
Glycogenic Hepatopathy
Glycogenic hepatopathy is a distinctive clinicopathologic entity where the normal balance between glycogenesis and glycogenolysis in hepatocytes is disrupted due to poor control of blood sugar levels. This leads to excess glycogen accumulation within hepatocyte cytoplasm.
CLINICAL FINDINGS. The classic clinical setting for the development of glycogenic hepatopathy is poor glycemic control in individuals with type 1 diabetes mellitus. Glycogenic hepatopathy can also be part of the Mauriac syndrome. This syndrome results from very poorly controlled type 1 diabetes and includes the findings of growth retardation, delayed puberty, cushingoid features, and hypercholesterolemia. These clinical findings are accompanied by hepatomegaly, abnormal liver enzymes, and glycogenic hepatopathy on liver biopsy. The Mauriac syndrome is only rarely seen today because of improved diagnosis and care of type 1 diabetes, but glycogenic hepatopathy is still seen on a regular basis.
Glycogenic hepatopathy is universally accompanied by elevated transaminase levels and hepatomegaly. In some cases, the patient’s hepatic enzymes can exceed 10 times the upper limit of normal.14 Also of note, the enzymes levels may fluctuate considerably over time.15 In all cases, the liver’s synthetic function is well preserved. Ascites is a dramatic but rare presentation of glycogenic hepatopathy14 and is often clinically misinterpreted as evidence for advanced liver disease. However, the ascites results from compression of the sinusoids by the rapidly expanding hepatocyte cytoplasm and typically resolves with adequate control of blood sugar.14,16 Also of note, both fatty liver disease and glycogenic hepatopathy can appear echogenic on ultrasound evaluation16,17 and individuals may have a working clinical diagnosis of fatty liver disease at the time of liver biopsy. The hepatomegaly and abnormal liver enzymes associated with glycogenic hepatopathy will improve with glycemic control.18,19 In addition, the histologic findings resolve with proper blood sugar control.20
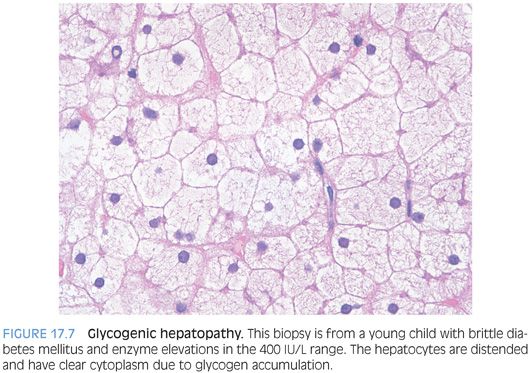
HISTOLOGIC FINDINGS. The hepatocytes have abundant pale cytoplasm, often with accentuation of the hepatocyte membranes (Fig. 17.7, eFig. 17.6). The distinctive histologic findings along with a history of diabetes are usually sufficient to make a diagnosis of glycogenic hepatopathy. If you would like, a periodic acid–Schiff (PAS) stain can be used to highlight the glycogen within the cytoplasm of hepatocytes, and the staining will disappear after digestion with diastase. However, remember that even normal livers will have abundant PAS positivity (eFig. 17.7) and a diagnosis of glycogenic hepatopathy requires the typical H&E findings and appropriate clinical setting.
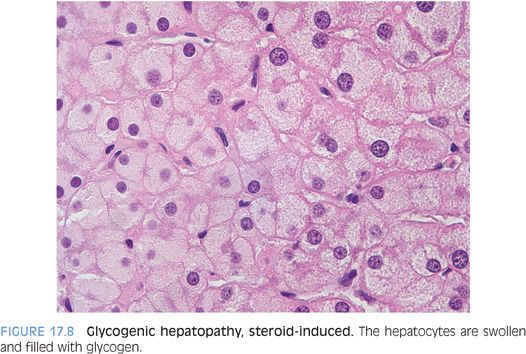
The differential for cases of histologically typical glycogenic hepatopathy includes medication effect. For example, short-term, high-dose steroid therapy can lead to glycogenic hepatopathy.21 In fact, the clinical presentation after high-dose steroid therapy (hepatomegaly and elevated transaminases elevations), as well as the histologic findings, can be very similar, although generally less striking than the changes of glycogenic hepatopathy in the setting of diabetes mellitus (Fig. 17.8). While counterintuitive, the differential also includes malnutrition, although the precise mechanism remains unclear.22 Glycogenic hepatopathy can also be induced by poorly controlled blood sugar levels in patients with type 2 diabetes.16 Finally, the differential also includes dumping syndrome secondary to fundoplication for gastroesophageal reflux disease.23
Adults with type 2 diabetes can have milder forms of liver glycogenosis that only become evident when biopsies are performed to evaluate the liver for other disease processes, such as chronic hepatitis C or fatty liver disease.16 The clinical and pathologic correlates of glycogenosis in this setting have not been well characterized to date, but the findings are patchy, in contrast to the diffuse findings in glycogenic hepatopathy, and this finding should not be confused with or diagnosed as glycogenic hepatopathy. Glycogen storage disease can also appear very similar to glycogenic hepatopathy histologically, but at the practical level, the clinical situations are sufficiently different and there is little difficulty in separating these two diagnoses.
Macrovesicular Steatosis
Although glycogenic hepatopathy is the most frequent histologic finding in patients with type 2 diabetes and hepatomegaly, fatty liver disease is also common. For example, in one study of 99 children with diabetes and hepatomegaly, glycogen accumulation was the most common cause of hepatomegaly. Moderate glycogen accumulation was seen in 22% of cases and marked glycogen accumulation in 19% of cases.24 However, fatty liver was also seen in nearly half of the total number of cases. Although the fatty change was usually mild, it did appear to explain the hepatomegaly in 8% of the children.24
Diabetic Hepatosclerosis
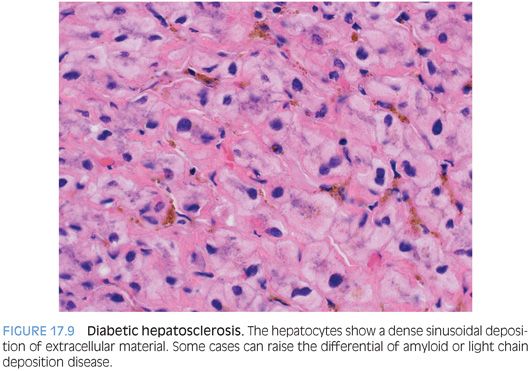
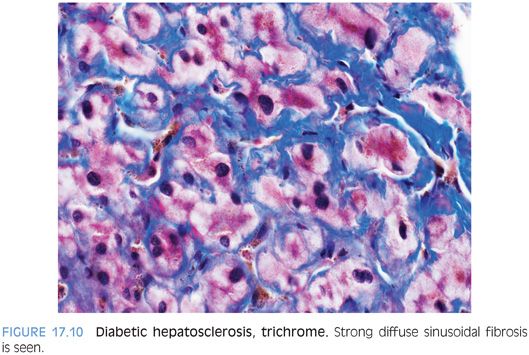
Recently, an additional pattern of liver injury, termed hepatosclerosis, has been described in a series of liver biopsies from 12 patients with diabetes.25 The liver biopsy specimens can show dense sinusoidal fibrosis, even though the livers are noncirrhotic (Fig. 17.9). A trichrome stain highlights the striking sinusoidal fibrosis (Fig. 17.10). Hepatosclerosis can be an independent finding that is not accompanied by fatty liver or by glycogenic hepatopathy. Affected individuals often have extensive histories of microangiopathic complications from their diabetes mellitus that involve multiple organ systems, suggesting that hepatosclerosis is a result of microangiopathic disease of the liver. Autopsy studies have found a frequency that ranges from 2% to 12% in patients with diabetes.23,26
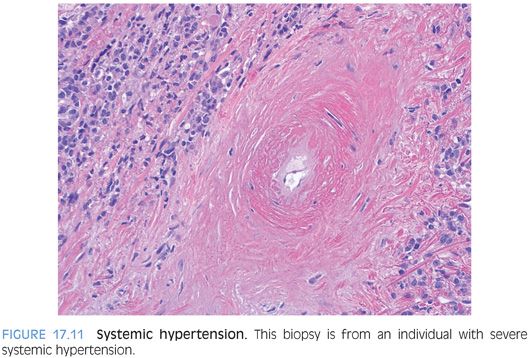
SYSTEMIC HYPERTENSION
Systemic hypertension–related changes are more commonly evident in the larger vessels and thus are more commonly seen in resection or autopsy specimens compared to biopsies. However, in some individuals with severe hypertension, the smaller arteries seen in a percutaneous liver biopsy can show significant intimal thickening and fibrosis (Fig. 17.11, eFig. 17.8). The changes can closely resemble amyloidosis, so it is helpful to perform a Congo red stain in these cases.