CHAPTER 86 Liver Disease Caused by Drugs
DEFINITIONS AND IMPORTANCE
The term drug-induced liver disease should be confined to cases in which the nature of liver injury has been characterized histologically. With the exception of acetaminophen, anticancer drugs, and some botanical or industrial hepatotoxins (see Chapter 87), most cases of drug-induced liver disease represent adverse drug reactions or hepatic drug reactions. These effects are noxious and unintentional and occur at doses recommended for prophylaxis or therapy. The latent period is longer (typically one week to three or six months) than that for direct hepatotoxins (hours to a few days), and extrahepatic features of drug hypersensitivity may be present.
Although drug-induced liver disease is a relatively uncommon cause of jaundice or acute hepatitis in the community, it is an important cause of more severe types of acute liver disease, particularly among older people (see Epidemiology). The overall mortality rate among patients hospitalized for drug-induced liver injury is approximately 10%1 but varies greatly for individual drugs.2,3 The reported frequencies of individual hepatic drug reactions are often underestimated because of the inadequacy of spontaneous reporting by physicians and pharmacists.2,3 With more reliable prospective and epidemiologic techniques, the frequency (or risk) of most types of drug-induced liver disease is between one per 10,000 and one per 100,000 persons exposed.4 Because these responses to drug exposure are clearly rare and unpredictable, they are often termed idiosyncratic drug reactions. Their rarity blunts diagnostic acumen because most clinicians will see few, if any, cases and therefore do not have an appropriate level of clinical suspicion. This concern applies especially to complementary and alternative medicine (CAM), as discussed in Chapter 87. Failure to withdraw the causative agent after the onset of symptoms of drug hepatitis or inadvertent reexposure to such a drug is a common and avoidable factor in acute liver failure attributable to drug-induced liver injury.5–8 Another challenge is that hepatic drug reactions produce an array of clinical syndromes and pathologic findings that mimic known hepatobiliary diseases. Furthermore, although individual agents (and some drug classes) typically produce a characteristic “signature syndrome,” they can also be associated with other and sometimes multiple clinicopathological syndromes.
Drug-induced liver injury is the most common reason for withdrawal of an approved drug from the market. The subject therefore has medicoeconomic, legal, and regulatory ramifications. Because of the low frequency of most types of idiosyncratic drug reactions that involve the liver, serious hepatotoxicity is not usually detected until post-marketing surveillance is conducted. Historically, drugs that have developed a reputation for potential hepatotoxicity usually have been replaced by more acceptable alternatives. Examples include troglitazone, the prototypic thiazolidinedione and bromfenac, a nonsteroidal anti-inflammatory drug (NSAID), both of which were withdrawn from the market because of several cases of fatal acute liver failure.5–9
The burgeoning number of available conventional medications and CAM preparations now includes many hundreds that can be cited as rare causes of drug-induced liver disease. This increasing number of potentially causative agents poses several challenges to clinicians,5–10 including concern about what constitutes an adequate level of patient information at the time a drug is prescribed and the reliability of evidence linking an individual agent to a particular type of liver injury.5,11–13 Another development is the appreciation that drug toxicity, in the context of complex medical situations, can interact with other causes of liver injury. Noteworthy examples of such situations are bone marrow transplantation; cancer chemotherapy; highly active antiretroviral therapy (HAART) for human immunodeficiency virus (HIV) infection and the acquired immunodeficiency syndrome (AIDS); use of antituberculosis drugs in patients with chronic viral hepatitis; rifampin hepatitis in patients with primary biliary cirrhosis (Chapter 89); nonalcoholic fatty liver disease (NAFLD)—particularly nonalcoholic steatohepatitis (NASH)—precipitated by tamoxifen; and possibly other drugs in overweight persons with type 2 diabetes mellitus and the metabolic syndrome.
EPIDEMIOLOGY
Frequency or risk—the number of adverse reactions for a given number of persons exposed—is the best term for expressing how common a drug reaction is. Time-dependent terms such as incidence and prevalence are not appropriate for drug reactions because the frequency is not linearly related to the duration of exposure. For most reactions, the onset occurs within a relatively short exposure time, or latent period, although some rarer types of chronic liver disease occur after many months or years. The frequency of drug-induced liver disease is usually based on the reported rate of drug reactions; such reports are usually a voluntary part of post-marketing surveillance and are submitted to pharmaceutical companies or adverse drug reaction monitoring bodies. In the United States, following approval by the U.S. Food and Drug Administration (FDA), pharmaceutical companies are required to report serious adverse events (any incident resulting in death, a threat to life, hospitalization, or permanent disability [Code of Federal Regulations]). Surveillance becomes a more passive process, however, when a drug is approved for marketing, and physicians and pharmacists are encouraged to file voluntary written reports through the MediWatch program. Similar systems operate in most industrialized countries. Nevertheless, MediWatch receives reports for fewer than 10% of adverse drug reactions,2 and in France fewer than 6% of hepatic adverse drug reactions are reported.3 The situation may be somewhat better in Sweden, but the annual reported incidence of adverse drug reactions of 2.2 per 100,000 in the population over the age of 15 is still much lower than the predicted incidence of 14 per 100,000.3 A prospective surveillance study in Spain measured the annual incidence of drug-related acute serious liver disease as 7.4 per million inhabitants.4
CASE DEFINITION: WHICH AGENT?
At least 300 agents have been implicated in drug-induced liver injury.10 The evidence for most drugs, however, is confined to individual or small numbers of case reports, especially in letters to scientific journals or to regulatory authorities, or small observational series. Therefore, for most agents, the evidence that they could cause liver injury is circumstantial and incomplete. Reports often lack pathologic definition, full exclusion of other disorders (for older reports), and logistic imputation of causality, especially with respect to temporal associations (see Diagnosis).5,9,10 Overall, probably fewer than 50 agents have been implicated reliably as causes of drug-induced liver disease. In general, agents used most commonly in clinical practice and in the community, including antimicrobials, antineoplastic agents, and NSAIDs, are those that have been implicated most often in drug-induced liver injury in larger series. The challenge of identifying the culprit drug among multiple candidates is discussed later.1,4,5,6,11
FREQUENCIES OF HEPATIC DRUG REACTIONS
Because of incomplete reporting, frequencies of hepatic drug reactions may often be underestimated. These estimated frequencies are also crude indicators of risk because of the inherent inaccuracies of case definitions (see Diagnosis)5,9,10 and because case recognition and reporting depend on the skill and motivation of observers. The increased interest of prescribers when initial cases of drug-induced liver disease have been described, together with inappropriate prescribing (e.g., prolonged use of bromfenac, which was approved only for seven days of use, and overprescribing of flucloxacillin and amoxicillin-clavulanic acid in some countries) can give rise to apparent “mini-epidemics.” More appropriate epidemiologic methods applied to hepatotoxicity have included prescription event monitoring, record linkage, and case-control studies. Prescription event monitoring and record linkage have been used to estimate the frequency of liver injury with some antimicrobials (erythromycins, sulfonamides, tetracyclines, flucloxacillin, amoxicillin-clavulanate) and NSAIDs.11
Epidemiologic studies confirm the rarity of drug-induced liver disease with currently used agents. For NSAIDs, the risk of liver injury is between 1 and 10 per 100,000 individuals exposed1,4,5,6; amoxicillin-clavulanic acid has been associated with cholestatic hepatitis in 1 to 2 per 100,000 exposed persons1,4,5,6,8; and low-dose tetracyclines have caused hepatotoxicity in less than one case per million persons exposed.1,4–6 The frequency of liver injury may be higher for agents that exert a metabolic type of hepatotoxicity. For example, isoniazid causes liver injury in up to 2% of persons exposed; the risk depends on the patient’s age and gender, concomitant exposure to other agents, and presence of hepatitis B virus (HBV) and possibly hepatitis C virus (HCV) infections.12 For some drugs in which other host factors play an etiopathogenic role, case-control studies have been used to define attributable risk. Examples include the implication of aspirin in Reye’s syndrome and oral contraceptives in liver tumors and hepatic vein thrombosis.
A relationship may exist between the frequency and severity of serum ALT elevations that indicate liver injury and the risk of severe hepatotoxicity. This relationship was proposed in the 1970s by the late Hyman Zimmerman.6 According to “Hy’s rule,” elevations of serum ALT levels to eight-fold or more above the upper limit of normal or associated increases in the serum bilirubin concentration indicate a potential for the drug to cause acute liver failure at a rate of about 10% of the number of cases of jaundice. Therefore, if two cases of jaundice associated with drug-induced liver injury are observed in a total phase 3 clinical trial experience of 2500 patients, approximately one case of acute liver failure would be expected for every 12,500 subjects who were prescribed the drug during the marketing phase.
IMPORTANCE OF DRUGS AS A CAUSE OF LIVER DISEASE
Hepatotoxicity accounts for less than 5% of cases of jaundice or acute hepatitis in the community and for even fewer cases of chronic liver disease5,6; however, drugs are an important cause of more severe types of liver disease and for liver disease in older people. They account for 10% of cases of severe hepatitis admitted to the hospital in France6 and for 43% of cases of hepatitis among patients 50 years of age or older.7 Drugs account for more than half of the cases of acute liver failure referred to special units in the United States7 and between 20% and 75% of cases of acute liver failure in other industrialized countries.4,7 The pattern of agents incriminated varies among countries; for example, herbal medicines are a relatively more common cause in Asian countries than in other countries (Chapter 87).
RISK FACTORS
For dose-dependent hepatotoxins such as acetaminophen and methotrexate and for some idiosyncratic reactions that are partly dependent on dose (e.g., bromfenac, tetracyclines, dantrolene, tacrine, oxypenicillins), the factors that influence the risk of drug-induced liver disease include the dose of the drug, blood level of the drug, and duration of intake. For idiosyncratic reactions, however, host determinants appear to be central to liver injury. The most critical determinant is likely to be genetic predisposition, but other “constitutional” and environmental factors can influence the risk of liver injury, as summarized in Table 86-1. The most important factors are age,4 gender, exposure to other substances, a history or family history of previous drug reactions, other risk factors for liver disease, and concomitant medical disorders.
Table 86-1 Factors Influencing the Risk of Liver Diseases Caused by Drugs
| FACTOR | EXAMPLES OF DRUGS AFFECTED | INFLUENCE |
|---|---|---|
| Age | Isoniazid, nitrofurantoin, halothane, troglitazone | Age >60 years: increased frequency, increased severity |
| Valproic acid, salicylates | More common in children | |
| Gender | Halothane, minocycline, nitrofurantoin | More common in women, especially with chronic hepatitis |
| Amoxicillin-clavulanic acid, azathioprine | More common in men | |
| Dose | Acetaminophen, aspirin; some herbal medicines | Blood levels are directly related to the risk of hepatotoxicity |
| Tetracycline, tacrine, oxypenicillins | Idiosyncratic reactions, but partial relationship to dose | |
| Methotrexate, vitamin A | Total dose, dosing frequency, duration of exposure is related to the risk of hepatic fibrosis | |
| Genetic factors | Halothane, phenytoin, sulfonamides | Multiple cases in families |
| Amoxicillin-clavulanic acid | Strong HLA association | |
| Valproic acid | Familial cases, association with mitochondrial enzyme deficiencies | |
| History of other drug reactions | Isoflurane, halothane, enflurane | Instances of cross-sensitivity have been reported among members of each drug class but are rare |
| Erythromycins | ||
| Diclofenac, ibuprofen, tiaprofenic acid | ||
| Sulfonamides, COX-2 inhibitors | ||
| Other drugs | Acetaminophen | Isoniazid, zidovudine, and phenytoin lower dose threshold and increase severity of hepatotoxicity |
| Valproic acid | Other antiepileptics increase risk of hepatotoxicity | |
| Anticancer drugs | Interactive vascular toxicity | |
| Excessive alcohol use | Acetaminophen hepatotoxicity | Lowered dose threshold, poorer outcome |
| Isoniazid, methotrexate | Increased risk of liver injury, hepatic fibrosis | |
| Nutritional status: | ||
| Obesity | Halothane, troglitazone, tamoxifen, methotrexate | Increased risk of liver injury; hepatic fibrosis |
| Fasting | Acetaminophen | Increased risk of hepatotoxicity |
| Preexisting liver disease | Hycanthone, pemoline | Increased risk of liver injury |
| Antituberculosis drugs, ibuprofen | Increased risk of liver injury with chronic hepatitis B and C | |
| Other diseases/conditions: | ||
| Diabetes mellitus | Methotrexate | Increased risk of hepatic fibrosis |
| HIV infection/AIDS | Sulfonamides | Increased risk of hypersensitivity |
| Renal failure | Tetracycline, methotrexate | Increased risk of liver injury, hepatic fibrosis |
| Organ transplantation | Azathioprine, thioguanine, busulfan | Increased risk of vascular toxicity |
AIDS, acquired immunodeficiency syndrome; COX-2, cyclooxygenase-2; HIV, human immunodeficiency virus; HLA, human leukocyte antigen.
Genetic Factors
Genetic determinants predispose to drug-induced liver disease,13 as they do for other types of drug reaction, such as penicillin allergy. Atopic patients have been thought to have an increased risk of some types of drug hepatitis, but this increase in risk has not been proved. Genetic factors determine the activity of drug-activating and antioxidant pathways, encode pathways of canalicular bile secretion, and modulate the immune response, tissue stress responses, and cell death pathways (see Chapter 72). Documented examples of drugs associated with a familial predisposition to adverse hepatic drug reactions are few and include valproic acid and phenytoin.5,8,13 Inherited mitochondrial diseases are a risk factor for valproic acid–induced hepatotoxicity.14 Some forms of drug-induced liver disease, particularly drug-induced hepatitis and granulomatous reactions, can be associated with the reactive metabolite syndrome (see later).
Weak associations have been reported between specific human leukocyte antigen (HLA) haplotypes and some types of drug-induced liver disease. Andrade and colleagues13 found positive associations between the class II HLA haplotype and cholestatic or mixed liver damage for some drugs. They suggested that no specific HLA allele predisposed to the overall risk of drug-induced liver disease but that the pattern of liver injury could be influenced by these genetic determinants. Other investigators have found stronger associations between the HLA haplotype and cholestatic reactions to amoxicillin-clavulanic acid and ticlopidine (see later).13,15
Age
Most hepatic drug reactions are more common in adults than in children. Exceptions include valproic acid hepatotoxicity, which is most common in infants younger than three years of age and rare in adults, and Reye’s syndrome, in which salicylates play a key role.16,17 As discussed later, both may be examples of mitochondrial toxicity.14 In adults, the risk of isoniazid-associated hepatotoxicity is greater in persons older than 40 years of age. Similar observations have been made for nitrofurantoin, halothane, etretinate, diclofenac, and troglitazone.1,4,5,10 The increased frequency of adverse drug reactions in older subjects is largely the result of increased exposure, the use of multiple agents, and altered drug disposition.1,4,5,10 In addition, clinical severity of hepatotoxicity increases strikingly with age, as exemplified by fatal reactions to isoniazid and halothane.
Gender
Women are particularly predisposed to drug-induced hepatitis, a difference that cannot be attributed simply to increased exposure. Examples include toxicity caused by halothane, nitrofurantoin, sulfonamides, flucloxacillin, minocycline, and troglitazone.5,6 Drug-induced chronic hepatitis caused by nitrofurantoin, diclofenac, or minocycline has an even more pronounced female preponderance.6 Conversely, equal sex frequency or even male preponderance is common for some drug reactions characterized by cholestasis, as for amoxicillin-clavulanic acid. Azathioprine-induced liver disease is more likely to develop in male renal transplant recipients than in female recipients.18
Concomitant Exposure to Other Agents
Patients who are taking multiple drugs are more likely to experience an adverse reaction than those who are taking one agent.5,9,10 The mechanisms include enhanced cytochrome P450 (CYP)-mediated metabolism of the second drug to a toxic intermediate (see later). Examples discussed later include toxicity caused by acetaminophen, isoniazid, valproic acid, other anticonvulsants, and anticancer drugs. Alternatively, drugs may alter the disposition of other agents by reducing bile flow or competing with canalicular pathways for biliary excretion (phase 3 drug elimination) (see later). This mechanism may account for apparent interactions between oral contraceptive steroids and other drugs to produce cholestasis. Drugs or their metabolites may also interact in mechanisms of cellular toxicity and cell death that involve mitochondrial injury, intracellular signaling pathways, activation of transcription factors, and regulation of hepatic genes involved in controlling the response to stress and injury that triggers pro-inflammatory and cell death processes.19,20
Previous Drug Reactions
A history of an adverse drug reaction generally increases the risk of reactions to the same drug and also to some other agents (see later). Nevertheless, instances of cross-sensitivity to related agents in cases of drug-induced liver disease are surprisingly uncommon. Examples of cross-sensitivity between drugs (or drug classes) include the haloalkane anesthetics (see Chapter 87), erythromycins, phenothiazines and tricyclic antidepressants, isoniazid and pyrazinamide, sulfonamides and other sulfur-containing compounds (e.g., some clyclooxygenase-2 [COX-2] inhibitors), and some NSAIDs. A crucial point is that a previous reaction to the same drug is a major risk factor for an increase in the severity of drug-induced liver injury.6
Nutritional Status
Obesity is strongly associated with the risk of halothane hepatitis (see Chapter 87) and appears to be an independent risk factor for NASH and hepatic fibrosis in persons taking methotrexate or tamoxifen. Fasting also predisposes to acetaminophen hepatotoxicity,21 and a role for undernutrition has been proposed in isoniazid hepatotoxicity.22
Preexisting Liver Disease
In general, liver diseases such as alcoholic cirrhosis and cholestasis do not predispose to adverse hepatic reactions. Exceptions include toxicity to some anticancer drugs, niacin, pemoline, and hycanthone. Preexisting liver disease is a critical determinant of methotrexate-induced hepatic fibrosis (discussed later). Patients with chronic HBV infection12 and possibly those with chronic HCV infection or HIV/AIDS appear to be at heightened risk of liver injury during anti-tuberculosis or HAART therapy,23 after exposure to ibuprofen and possibly other NSAIDs, after myeloablative therapy in preparation for bone marrow transplantation (resulting in sinusoidal obstruction syndrome [see later]),24 and possibly after taking antiandrogens, such as flutamide and cyproterone acetate.25 A particularly strong association has been observed between HCV infection (present in 33% of patients with HIV/AIDS) and the risk of liver injury during HAART; the risk may be increased 2- to 10-fold.26–30
Other Diseases
Rheumatoid arthritis appears to increase the risk of salicylate hepatotoxicity, and a curious, unexplained observation is that hepatitis associated with sulfasalazine appears to be more common in patients with rheumatoid arthritis than in those with inflammatory bowel disease.8–10,31,32 Diabetes mellitus, obesity, and chronic kidney disease predispose to methotrexate-induced hepatic fibrosis, whereas HIV/AIDS confers a heightened risk of sulfonamide hypersensitivity.31–33 A retrospective cohort study of five health maintenance organizations found that the age- and sex-standardized incidence of drug-induced acute liver failure in patients with diabetes mellitus was 0.08 to 0.15 per 1000 person-years, irrespective of the therapeutic agent used (the number using troglitazone was small); the incidence was highest (approximately 0.3 per 1000) during the first six months of exposure.32 Renal transplantation is a risk factor for azathioprine-associated vascular injury, whereas kidney disease predisposes to tetracycline-induced fatty liver.6 Finally, sinusoidal obstruction syndrome induced by anticancer drugs is more common after bone marrow transplantation24 and in persons with HCV infection.5,6,8–10,26
PATHOPHYSIOLOGY
PATHWAYS OF DRUG METABOLISM
As reviewed elsewhere,5,34 phase 1 pathways of drug metabolism include oxidation, reduction, and hydrolytic reactions. The products can be readily conjugated or excreted without further modification.
Cytochrome P450
Most type 1 reactions are catalyzed by microsomal drug oxidases, the key component of which is a hemoprotein of the CYP gene superfamily. The apparent promiscuity of drug oxidases toward drugs, environmental toxins, steroid hormones, lipids, and bile acids results from the existence of multiple closely related CYP proteins. More than 20 CYP enzymes are present in the human liver.34
The reaction cycle involves binding of molecular oxygen to the iron in the heme prosthetic group, with subsequent reduction of oxygen by acceptance of an electron from nicotinamide-adenine dinucleotide phosphate (NADPH) cytochrome P450 reductase, a flavoprotein reductase. The resulting “activated oxygen” is incorporated into the drug or another lipophilic compound. Reduction of oxygen and insertion into a drug substrate (“mixed function oxidation”) can result in formation of chemically reactive intermediates, including free radicals, electrophilic “oxy-intermediates” (e.g., unstable epoxides, quinone imines), and reduced (and therefore reactive) oxygen species (ROS). The quintessential example is the CYP2E1-catalyzed metabolite of acetaminophen, N-acetyl-p-benzoquinone imine (NAPQI), an oxidizing and arylating metabolite that is responsible for liver injury associated with acetaminophen hepatotoxicity. Other quinone compounds are potential reactive metabolites of troglitazone, quinine, and methyldopa. Epoxide metabolites of diterpenoids may be hepatotoxic products of the hepatic metabolism of some plant toxins (see Chapter 87).35 ROS have broad significance in the production of tissue injury, particularly by contributing to the production of oxidative stress and triggering tissue stress responses and cell death pathways, as discussed later.
The hepatic content of CYP proteins is higher in acinar zone 3 (see Chapter 71). Localization of CYP2E1 is usually confined to a narrow rim of hepatocytes 1 to 2 cells thick around the terminal hepatic venule. This finding explains in part the zonality of hepatic lesions produced by drugs and toxins, such as acetaminophen and carbon tetrachloride, which are converted to reactive metabolites.
Genetic and Environmental Determinants of Cytochrome P450 Enzymes
Pharmacogenetics and Polymorphisms of Cytochrome P450 Expression
The hepatic expression of each CYP enzyme is genetically determined. This finding largely explains the four-fold or greater differences in rates of drug metabolism among healthy subjects. Some CYPs, particularly minor forms, are also subject to polymorphic inheritance; therefore, occasional persons completely lack the encoded protein.34 One example is CYP2D6, the enzyme responsible for the metabolism of debrisoquine and perhexiline. Poor metabolizers lack CYP2D6 and accumulate perhexiline when given usual doses; lack of CYP2D6 is the critical determinant in serious adverse effects of perhexiline, including chronic hepatitis and cirrhosis.36 Other examples include CYPs 2C9 and 2C19, which affect the metabolism of S-warfarin, omeprazole, tolbutamide, and phenytoin and of S-mephenytoin, respectively34; 3% of white populations and 15% of Asians are poor metabolizers of S-mephenytoin.
Nutrition and Disease-Related Changes
A person’s nutritional status influences the expression of certain CYPs, both in health and with liver disease.5,10,20,34 Expression of CYP2E1 is increased by obesity, high fat intake, and fasting.20,34 Diseases that alter the expression of hepatic CYPs include diabetes mellitus (increased CYP2E1), hypothyroidism (decreased CYP1A), and hypopituitarism (decreased CYP3A4).34 Cirrhosis is associated with decreased levels of total cytochrome P450 and also with reduced hepatic perfusion; the result is a decrease in the clearance of drugs such as propranolol that are metabolized rapidly by the liver.34 The effects of cirrhosis vary, however, among individual CYP families (e.g., CYP1A levels are lowered, but CYP2C and CYP2D6 levels often are preserved) and with the type of liver disease (e.g., CYP3A4 levels are preserved with cholestatic liver disease but lowered with hepatocellular liver disease).
Adaptive Response and Enzyme Induction
Exposure to lipophilic substances results in an adaptive response that usually involves synthesis of new enzyme protein, a process termed enzyme induction. The molecular basis for genetic regulation of constitutive and inducible expression of the major human hepatic cytochrome P450, CYP3A4, has been determined.37 Agents such as rifampin interact with the pregnane X-receptor (PXR), a member of the orphan nuclear receptor family of transcriptional regulators.37 Activated PXR and the analogous constitutive androstane receptor (CAR) in turn bind to cognate nucleotide sequences upstream to the CYP3A4 structural gene within a “xenobiotic-regulatory enhancer module” (XREM). This interaction regulates the CYP3A4 promoter downstream and ultimately the transcription of CYP3A4 protein. Similar control mechanisms apply to several other CYP pathways,37,38 particularly those involved with bile acid synthesis in which the nuclear receptors implicated include the farnesoid X-receptor (FXR), which down-regulates bile acid synthesis and up-regulates bile salt excretory pathways, and liver X receptor, a positive regulator of bile acid synthesis via CYP7A (see also Chapter 64).37
Common examples of the induction of microsomal enzymes by environmental compounds include the effect of smoking cigarettes and cannabis on CYP1A238 and of alcohol on CYP2E1 and possibly CYP3A4.39 Several drugs are potent inducers of CYP enzymes. Isoniazid induces CYP2E1, whereas phenobarbital and phenytoin increase the expression of multiple CYPs.34 Rifampin is a potent inducer of CYP3A4, as is hypericum,40 the active ingredient of St John’s wort, a commonly used herbal medicine, thereby causing interactions between conventional medicines and a CAM preparation. Further descriptions of the regulation of hepatic drug metabolizing enzymes have been published elsewhere.34,38
Phase 3 Pathways
Several transporters secrete drugs, drug metabolites, or their conjugates into bile, and this mechanism is often referred to as phase 3 of hepatic drug elimination. These pathways involve ATP-binding cassette (ABC) proteins, which derive the energy for their transport functions from hydrolysis of ATP. ABC transport proteins are widely distributed in nature and include the cystic fibrosis transmembrane conductance regulator (CFTR) (see Chapter 76) and the canalicular and intestinal copper transporters (see Chapter 75). The role of ABC transport proteins in secretion of bile has been reviewed (see Chapter 64).37,38,41,42
Multidrug resistance protein 1 (MDR1) is highly expressed on the apical (canalicular) plasma membrane of hepatocytes, where it transports cationic drugs, particularly anticancer agents, into bile. Another family of ABC transporters, the multidrug resistance-associated proteins (MRPs), is also expressed in the liver. At least two members of this family excrete drug (and other) conjugates from hepatocytes: MRP-3 on the basolateral surface facilitates passage of drug conjugate into the sinusoidal circulation, and MRP-2, expressed on the canalicular membrane, pumps endogenous compounds (e.g., bilirubin diglucuronide, leukotriene-glutathionyl conjugates, glutathione) and drug conjugates into bile. The bile salt export pump (BSEP) and MDR3 (in humans) and Mdr2 (in mice) are other canalicular transporters concerned, respectively, with bile acid and phospholipid secretion into bile. Genetic polymorphisms of these genes are associated with human cholestatic liver diseases (see Chapters 64 and 76). BSEP interacts with several drugs.42
Regulation of the membrane expression and activity of these drug elimination pathways is complex. The possibility that their altered expression or impaired activity (by competition between agents, changes in membrane lipid composition, or damage from reactive metabolites or covalent binding) could lead to drug accumulation, impairment of bile flow, or cholestatic liver injury has been demonstrated for estrogens,43,44 troglitazone,45 terbinafine,46 and flucloxacillin47 and may have wider mechanistic importance for drug-induced cholestasis and other forms of liver injury.42
TOXIC MECHANISMS OF LIVER INJURY
Oxidative Stress and the Glutathione System
The liver is exposed to oxidative stress by the propensity of hepatocytes to reduce oxygen, particularly in mitochondria and also in microsomal electron transport systems (such as CYP2E1), and by NADPH-oxidase-catalyzed formation of ROS and nitroradicals in Kupffer cells, endothelial cells, and stimulated polymorphs and macrophages. To combat oxidative stress, the liver is well-endowed with antioxidant mechanisms, including micronutrients, such as vitamin E and vitamin C, thiol-rich proteins (e.g., metallothionein, ubiquinone), metal-sequestering proteins (e.g., ferritin), and enzymes that metabolize reactive metabolites (e.g., epoxide hydrolases), ROS (e.g., catalase, superoxide dismutase), and lipid peroxides (e.g., glutathione peroxidases). Glutathione (l-gamma-glutamyl-l-cyteine-glycine) is the most important antioxidant in the mammalian liver.19
Hepatocytes are the exclusive site of glutathione synthesis. Hepatic levels of glutathione are high (5 to 10 mmol/L) and can be increased by enhancing the supply of cysteine for glutathione synthesis; this mechanism is the cornerstone of thiol antidote therapy for acetaminophen poisoning. Hepatocyte glutathione synthesis increases in response to pro-oxidants, as occurs when CYP2E1 is overexpressed as a result of signaling via the redox-sensitive transcription factor Nrf.19,20,48,49 Glutathione is a critical cofactor for several antioxidant pathways, including thiol-disulfide exchange reactions and glutathione peroxidase. Glutathione peroxidase has a higher affinity for hydrogen peroxide than does catalase, and it disposes of lipid peroxides, free radicals, and electrophilic drug metabolites. Reduced glutathione is a cofactor for conjugation reactions catalyzed by the glutathione S-transferases. Other reactions proceed nonenzymatically. In turn, the products include glutathione-protein mixed disulfides and oxidized glutathione. The latter can be converted back to glutathione by proton donation catalyzed by glutathione reductase.
Normally, most glutathione within the hepatocyte is in the reduced state, indicating the importance of this pathway for maintenance of the redox capacity of the cell. The reduced form of NADPH is an essential cofactor for glutathione reductase; NADPH formation requires ATP, thereby illustrating a critical link between the energy-generating capacity of the liver and its ability to withstand oxidative stress. Glutathione is also compartmentalized within the hepatocyte, with the highest concentrations found in the cytosol. Adequate levels of glutathione are essential in mitochondria, where ROS are constantly being formed as a minor by-product of oxidative respiration and in response to some drugs or metabolites that interfere with the mitochondrial respiratory chain. Mitochondrial glutathione is maintained by active uptake from the cytosol, a transport system that is altered by chronic ethanol exposure, and is, therefore, another potential target of drug toxicity.19
Biochemical Mechanisms of Cellular Injury
Mechanisms once thought to be central to hepatotoxicity, such as covalent binding to cellular enzymes and peroxidation of membrane lipids, are no longer regarded as exclusive pathways of cellular damage. Rather, oxidation of proteins, phospholipid fatty acyl side chains (lipid peroxidation), and nucleosides appear to be components of the biochemical stress that characterizes toxic liver injury. Secondary reactions also may play a role; these reactions include post-translational modification of proteins via adenosine diphosphate (ADP) ribosylation or protease activation, cleavage of DNA by activation of endogenous endonucleases, and disruption of lipid membranes by activated phospholipases.20 Some of these catabolic reactions could be initiated by a rise in the cytosolic ionic calcium concentration [Ca2+]i, as a result of increased Ca2+ entry or release from internal stores in the endoplasmic reticulum and mitochondria.19,20
Types of Cell Death
Apoptosis
Apoptosis is an energy-dependent, genetically programmed form of cell death that typically results in controlled deletion of individual cells. In addition to its major roles in developmental biology, tissue regulation, and carcinogenesis, apoptosis is important in toxic, viral, and immune-mediated liver injury.50–53 The ultrastructural features of apoptosis are cell and nuclear shrinkage, condensation and margination of nuclear chromatin, plasma membrane blebbing, and ultimately fragmentation of the cell into membrane-bound bodies that contain intact mitochondria and other organelles. Engulfment of these apoptotic bodies by surrounding epithelial and mesenchymal cells conserves cell fragments that contain nucleic acid and intact mitochondria. These fragments are then digested by lysosomes and recycled without release of bioactive substances. As a consequence, apoptosis in it purest form (usually found only in vitro) does not incite an inflammatory tissue reaction. The cellular processes that occur in apoptosis are often mediated by caspases, a family of proteolytic enzymes that contain a cysteine at their active site and cleave polypeptides at aspartate residues; non–caspase-mediated programmed cell death has also been described in experimental hepatotoxicity (see also Chapter 72).
Apoptosis rarely, if ever, is the sole form of cell death in common forms of liver injury, such as ischemia-reperfusion injury, cholestasis, and toxic liver injury, all of which are typically associated with a hepatic inflammatory response. Whether or not activation of pro-death signals causes cell death depends on several factors, including pro-survival signals, the rapidity of the process, the availability of glutathione and ATP, and the role of other cell types. Some of these issues are discussed briefly here and are reviewed in more detail elsewhere.20,50–53
Mitochondria play a pivotal role in pathways that provoke or oppose apoptosis.50,51,53 In the external pathway, activation of the death domain of pro-apoptotic receptors recruits adapter molecules—Fas-associated death domain (FADD) and TNF receptor-associated death domain (TRADD)—which bind and activate procaspase 8 to form the death-inducing signaling complex (DISC). In turn, caspase 8 cleaves Bid, a pro-apoptotic member of the B cell lymphoma/leukemia (Bcl-2) family, to tBid. tBid causes translocation of Bax to the mitochondria, where it aggregates with Bak to promote permeability of the mitochondria.50 Release of cytochrome c and other pro-death molecules, including Smac (which binds caspase inhibitor proteins, such as inhibitor of apoptosis proteins [IAPs]) and apoptosis-inducing factor (AIF, also known as Apaf)51 allows formation of the “aptosome,” which activates caspase 9 and eventually caspase 3 to execute cell death (Fig. 86-1). Intracellular stresses in various sites release other mitochondrial permeabilizing proteins (e.g., Bmf from the cytoskeleton and Bim from the endoplasmic reticulum), whereas members of the Bcl-2 family, Bcl-2 and Bcl-XL, antagonize apoptosis and serve as survival factors by regulating the integrity of mitochondria; the protective mechanism is poorly understood. Stress-activated protein kinases, particularly c-jun N-terminal kinase (JNK) may also be pro-apoptotic52 by phosphorylating and inactivating the mitochondrial protective protein Bcl-XL.
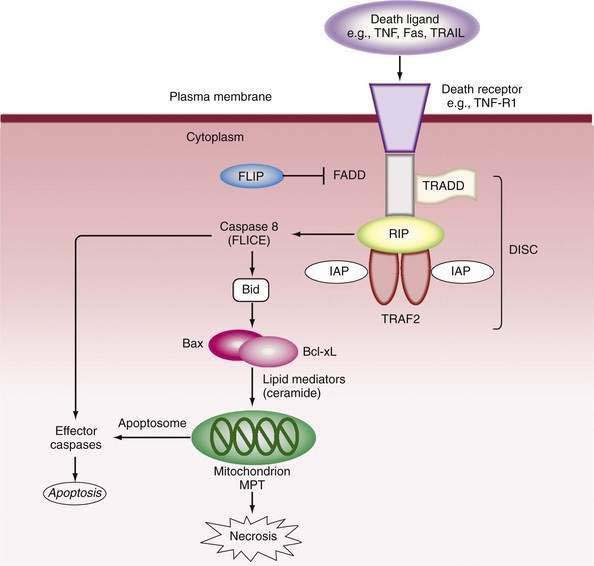
Execution of cell death by apoptosis usually occurs via activation of caspase 3, but more than one caspase-independent pathway of programmed cell death has been described.53 Stresses to the endoplasmic reticulum can bypass mitochondrial events by activation of caspase 12, which in turn activates caspase 9 independently of the apoptosome. The final steps of programmed cell death are energy dependent. Therefore, depletion of ATP abrogates the controlled attempt at “cell suicide,” resulting instead in necrosis (see later) or an overlapping pattern that has been designated as “apoptotic necrosis” or “necraptosis.”54,55 Furthermore, when apoptosis is massive, the capacity for rapid phagocytosis can be exceeded, and “secondary” necrosis can occur.55






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


