CHAPTER 73 Liver Chemistry and Function Tests
BILIRUBIN (see Chapter 20)
BILIRUBIN METABOLISM
Bilirubin is a breakdown product of heme (ferroprotoporphyrin IX). About 4 mg/kg body weight of bilirubin is produced each day, nearly 80% from the breakdown of hemoglobin in senescent red blood cells and prematurely destroyed erythroid cells in the bone marrow and the remainder from the turnover of hemoproteins such as myoglobin and cytochromes distributed throughout the body.1 The initial steps of bilirubin metabolism occur in reticuloendothelial cells, predominantly in the spleen. Heme is converted to biliverdin by the microsomal enzyme heme oxygenase. Biliverdin is then converted to bilirubin by the cytosolic enzyme biliverdin reductase.
After entering the hepatocyte, unconjugated bilirubin is bound in the cytosol to a number of proteins, including proteins in the glutathione S-transferase superfamily.2 These proteins serve to reduce efflux of bilirubin back into the serum and present the bilirubin for conjugation. The enzyme uridine-5′-diphosphate (UDP) glucuronyl transferase found in the endoplasmic reticulum solubilizes bilirubin by conjugating it to glucuronic acid to produce bilirubin monoglucuronide and diglucuronide.3 The now hydrophilic bilirubin diffuses to the canalicular membrane for excretion into the bile canaliculi. Conjugated bilirubin is transported across the canalicular membrane by the multiple drug resistance-associated protein 2 (MRP2) via an adenosine triphosphate (ATP)-dependent process.4 This is the only energy-dependent step in bilirubin metabolism and explains why even patients with fulminant hepatic failure have a predominantly conjugated hyperbilirubinemia. Once in the bile, conjugated bilirubin passes undisturbed until it reaches the distal ileum and colon, where bacteria containing β-glucuronidases hydrolyze conjugated bilirubin to unconjugated bilirubin, which is further reduced by bacteria to colorless urobilinogen.5 The urobilinogen is either excreted unchanged, oxidized and excreted as urobilin, which has an orange color, or absorbed passively by the intestine into the portal system as urobilinogen. The majority of the absorbed urobilinogen is re-excreted by the liver. A small percentage filters across the renal glomerulus and is excreted in urine. Unconjugated bilirubin is never found in urine because in the serum it is bound to albumin and not filtered by the glomerulus. The presence of bilirubin in urine indicates a conjugated hyperbilirubinemia and hepatobiliary disease.
MEASUREMENT OF SERUM BILIRUBIN
The terms direct and indirect bilirubin, which correspond roughly to conjugated and unconjugated bilirubin, respectively, derive from the original van den Bergh reaction.6 Serum bilirubin is still measured in clinical laboratories by some modification of this diazo reaction.7 In this assay, bilirubin is exposed to diazotized sulfanilic acid. The conjugated fraction of bilirubin reacts promptly, or “directly,” with the diazo reagent without the need for an accelerant and thereby allows measurement of the conjugated bilirubin fraction by photometric analysis within 30 to 60 seconds. The total bilirubin is measured 30 to 60 minutes after the addition of an accelerant such as alcohol or caffeine. The unconjugated, or indirect, fraction is then determined by subtracting the direct component from the total bilirubin.
Using the diazo method, normal values of total serum bilirubin are between 1.0 and 1.5 mg/dL, with 95% of a normal population falling between 0.2 and 0.9 mg/dL.8 Normal values for the indirect component are between 0.8 and 1.2 mg/dL. The diazo method, however, tends to overestimate the amount of conjugated bilirubin, particularly within the normal range. As a result, “normal” ranges for conjugated bilirubin have crept upward over time. In general, if the direct acting fraction is less than 15% of the total, the bilirubin can be considered to be entirely indirect. The most frequently reported upper limit of normal for conjugated bilirubin is 0.3 mg/dL. The presence of even a mild increase in conjugated bilirubin in the serum should raise the possibility of liver injury. The measurement and fractionation of serum bilirubin in patients with jaundice does not allow differentiation between parenchymal (hepatocellular) and obstructive (cholestatic) jaundice.
The magnitude and duration of hyperbilirubinemia have not been critically assessed as prognostic markers. In general, the higher the serum bilirubin level in patients with viral hepatitis, the greater the hepatocellular damage and the longer the course of disease. Patients may die, however, of acute liver failure with only a modest elevation of serum bilirubin. Total serum bilirubin correlates with poor outcomes in alcoholic hepatitis and is a critical component of the model for end-stage liver disease (MELD) score, which is used to estimate survival of patients with end-stage liver disease (see later and Chapter 95).
APPROACH TO THE PATIENT WITH AN ELEVATED BILIRUBIN LEVEL
The evaluation of the patient with an isolated elevation of the serum bilirubin level is quite different from that of the patient with an elevated bilirubin associated with elevated liver enzyme levels; the latter suggests either a hepatocellular or cholestatic process, as discussed later. The first step in the evaluation of a patient with an isolated elevation of the serum bilirubin level is to fractionate the bilirubin to determine if it is conjugated or unconjugated bilirubin (Fig. 73-1). If less than 15% of the total is conjugated, one can be assured that virtually all the serum bilirubin is unconjugated. An overproduction of bilirubin as a result of excessive breakdown of hemoglobin can occur with any of a number of inherited or acquired disorders (Table 73-1). The patient’s medication history should be reviewed for drugs that can cause impaired hepatocellular uptake of bilirubin. If no cause is identified, a genetic enzyme deficiency that results in impaired conjugation of bilirubin, the most common of which is Gilbert’s syndrome, is likely.
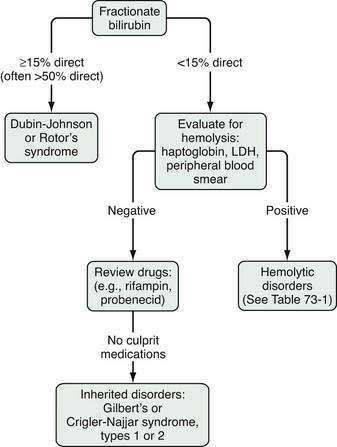
Figure 73-1. Evaluation of an isolated elevation of the serum bilirubin level. LDH, lactate dehydrogenase.
Table 73-1 Causes of Isolated Hyperbilirubinemia in Adults
| CAUSE | MECHANISM |
|---|---|
| Indirect Hyperbilirubinemia | |
| Hemolytic Disorders | Overproduction of bilirubin |
| Inherited | |
| Red cell enzyme defects (e.g., glucose-6-phosphate dehydrogenase deficiency) | |
| Sickle cell disease | |
| Spherocytosis and elliptocytosis | |
| Acquired | |
| Drugs and toxins | |
| Hypersplenism | |
| Immune mediated | |
| Paroxysmal nocturnal hemoglobinuria | |
| Traumatic: macro- or microvascular injury | |
| Ineffective Erythropoiesis | Overproduction of bilirubin |
| Cobalamin deficiency | |
| Folate deficiency | |
| Profound iron deficiency | |
| Thalassemia | |
| Drugs: Rifampin, Probenecid | Impaired hepatocellular uptake |
| Inherited Conditions Crigler-Najjar syndrome types I and II | Impaired conjugation of bilirubin |
| Gilbert’s syndrome | |
| Other | |
| Hematoma | Overproduction of bilirubin |
| Direct Hyperbilirubinemia | |
| Inherited Conditions | |
| Dubin-Johnson syndrome Rotor’s syndrome | Impaired excretion of conjugated bilirubin |
As discussed in Chapter 20, Gilbert’s syndrome is common, with a reported incidence of 6% to 12% (see Table 20-2). A mutation in the TATAA element in the 5′ promoter region of the UDP glucuronyl transferase gene results in a reduction in enzyme activity to approximately one third of normal. The mildly elevated indirect serum hyperbilirubinemia seen in Gilbert’s syndrome is of no clinical consequence. This benign clinical course contrasts with those of much rarer conditions, Crigler-Najjar syndrome, types I and II (see Table 20-2). The mutations in these conditions result in significantly reduced UDP glucuronyl transferase activity: <10% in Crigler-Najjar type II and complete absence of enzyme activity in Crigler-Najjar type I, resulting in much greater elevations of unconjugated serum bilirubin to levels that carry an increased risk of kernicterus.
When an isolated hyperbilirubinemia is associated with a conjugated fraction of >15%, and typically >50%, the diagnosis is either the uncommon Dubin-Johnson syndrome or the even rarer Rotor’s syndrome (see Fig. 73-1, Table 20-2, and Table 64-4). The defect in Dubin-Johnson syndrome is in the MRP2 gene. The defect in Rotor’s syndrome has yet to be defined, but in both syndromes excretion of conjugated bilirubin across the bile canalicular membrane is reduced, resulting in an increased conjugated serum bilirubin level. Neither syndrome is associated with adverse clinical outcomes. Additional genetic disorders of bile acid transport that may be associated with hyperbilirubinemia are discussed in Chapters 64 and 76.
AMINOTRANSFERASES
The serum aminotransferases (also called transaminases), the most sensitive markers of acute hepatocellular injury, have been used to identify liver disease since the 1950s.9 ALT (formerly serum glutamic pyruvic transaminase, or SGPT) and AST (formerly serum glutamic oxaloacetic transaminase, or SGOT) catalyze the transfer of the α-amino groups of alanine and l-aspartic acid, respectively, to the α-keto group of ketoglutaric acid. AST, found in cytosol and mitochondria, is widely distributed throughout the body; it is found, in order of decreasing concentration, in liver, cardiac muscle, skeletal muscle, kidney, brain, pancreas, lung, leukocytes, and erythrocytes. ALT, a cytosolic enzyme also found in many organs, is present in greatest concentration by far in the liver and is, therefore, a more specific indicator of liver injury. Increases in serum values of the aminotransferases reflect either damage to tissues rich in these enzymes or changes in cell membrane permeability that allow ALT and AST to leak into serum; hepatocyte necrosis is not required for the release of aminotransferases, and the degree of elevation of the aminotransferases does not correlate with the extent of liver injury.10
Aminotransferases have no function in serum and act like other serum proteins. They are distributed in plasma and interstitial fluid and have half-lives measured in days. The activity of ALT and AST at any moment reflects the relative rate at which they enter and leave the circulation. They are probably cleared by cells in the reticuloendothelial system, with AST cleared more rapidly than ALT. Normal values for aminotransferases in serum vary widely among laboratories, but values gaining general acceptance are <30 U/L for men and <19 U/L for women. The inter-laboratory variation in the normal range is the result of technical issues; no reference standards exist to establish the upper limits of normal for serum ALT and AST levels. Therefore, each reference laboratory is responsible for identifying a locally defined reference population or for using a normal range first established in the 1950s.9 The normal range is defined as the mean of the reference population plus 2 standard deviations; approximately 95% of a uniformly distributed population will fall within this “normal” range. Some investigators have recommended revisions of normal values for the aminotransferases with adjustments for sex and body mass index, but others have raised concern about the potential costs and unclear benefits of implementing such a change.11–15
APPROACH TO THE PATIENT WITH AN ELEVATED AMINOTRANSFERASE LEVEL
Serum aminotransferase levels are typically elevated in all forms of liver injury; levels up to 300 U/L are nonspecific. In certain circumstances the degree and pattern of elevation of the aminotransferases, evaluated in the context of a patient’s characteristics, symptoms, and physical examination findings, can suggest particular diagnoses and direct the subsequent evaluation (Table 73-2). The differential diagnosis of marked elevations of aminotransferase levels (>1000 U/L) includes viral hepatitis (A to E), toxin or drug-induced liver injury, ischemic hepatitis, and less commonly, autoimmune hepatitis, acute Budd-Chiari syndrome, fulminant Wilson disease, and acute obstruction of the biliary tract.
Table 73-2 Causes of Elevated Serum Aminotransferase Levels*
| Chronic, Mild Elevations, ALT > AST (<150 U/L or 5 × normal) |
| Hepatic Causes |
| Nonhepatic Causes |
| Severe, Acute Elevations, ALT > AST (>1000 U/L or >20-25 × normal) |
| Hepatic Causes |
| Severe, Acute Elevations, AST > ALT (>1000 U/L or >20-25 × normal) |
| Hepatic Cause |
| Medications or toxins in a patient with underlying alcoholic liver injury |
| Nonhepatic Cause |
| Acute rhabdomyolysis |
| Chronic, Mild Elevations, AST > ALT (<150 U/L, <5 × normal) |
| Hepatic Causes |
| Nonhepatic Causes |
ALT, alanine aminotransferase; AST, aspartate aminotransferase.
* Virtually any liver disease can cause moderate aminotransferase elevations (5-15 × normal)
The ratio of AST to ALT in serum is helpful in a few specific circumstances—perhaps most importantly in the recognition of alcoholic liver disease. If the AST level is less than 300 U/L, a ratio of AST to ALT of more than 2 suggests alcoholic liver disease, and a ratio of more than 3 is highly suggestive of alcoholic liver disease.16 The ratio results from a deficiency of pyridoxal 5′-phosphate in patients with alcoholic liver disease; ALT synthesis in the liver requires pyridoxal phosphate more than does AST synthesis.17 When a patient with chronic alcoholic liver disease sustains a superimposed liver injury, particularly acetaminophen hepatotoxicity, the aminotransferase level can be strikingly elevated, yet the AST/ALT ratio is maintained.
An increased ratio of AST to ALT may also be seen in muscle disorders. The degree of elevation is typically less than 300 U/L, but in rare cases, such as rhabdomyolysis, levels observed in patients with acute hepatocellular disease can be reached. In cases of acute muscle injury, the AST/ALT ratio may initially be greater than 3 : 1, but the ratio quickly declines toward 1 : 1 because of the shorter serum half-life of AST.18 The ratio typically is close to 1 : 1 in patients with chronic muscle diseases.
Although the AST to ALT ratio is typically less than 1 in patients with chronic viral hepatitis and nonalcoholic fatty liver disease (NAFLD), a number of investigators have observed that, as cirrhosis develops, the ratio rises and may become greater than 1. Studies have shown that an AST/ALT ratio of greater than 1 as an indicator of cirrhosis in patients with chronic hepatitis C has a high specificity (94% to 100%) but a relatively low sensitivity (44% to 75%).19 The increase in AST/ALT ratio with the development of cirrhosis is believed to result from impaired functional hepatic blood flow, with a consequent decrease in hepatic sinusoidal uptake of AST.20
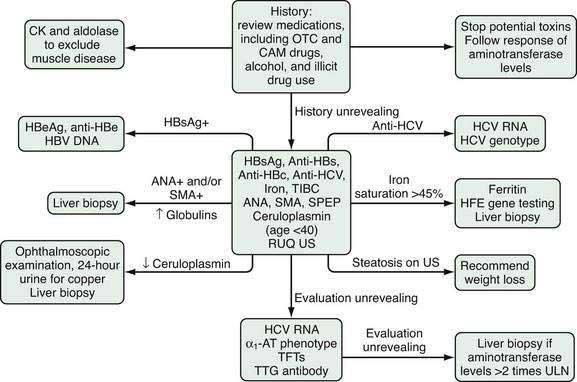
The majority of patients evaluated for elevated serum aminotransferase levels are asymptomatic and have mild elevations (≤5-fold) identified during routine screening. The first step in the evaluation of mildly elevated serum aminotransferase levels is to repeat the test to confirm persistence of the elevated value. If the aminotransferase level remains elevated, the recommended evaluation is illustrated in Figure 73-2. The initial step is to take a careful history focused on identifying all of the patient’s medications, including over-the-counter (OTC) medications, complementary and alternative medications (CAM), and substances of abuse. Correlating the use of medications temporally with the laboratory abnormalities will sometimes reveal a specific culprit. Almost any medication, including OTC medications, CAM, and substances of abuse, has the potential to elevate serum aminotransferase levels. Relatively common offending agents include nonsteroidal anti-inflammatory drugs, antibiotics, hydroxymethylglutaryl-coenzyme A reductase inhibitors, antiepileptics, and antituberculous medications (see Chapter 86). The association between use of a medication and liver enzyme elevations is readily established by stopping the medication and observing return of the enzyme levels to normal. Re-challenge with the suspect medication followed by a rise in serum aminotransferase levels is confirmatory but often not undertaken. Muscle disease should also be excluded by obtaining serum creatine kinase and aldolase levels.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


