Iron Absorption
Most iron is absorbed in the duodenum and proximal jejunum. Heme iron is disassociated from globin and then taken up by the enterocytes. Dietary non-heme iron requires additional steps and first has to be reduced from a ferric to a ferrous state before it can be transported into the cytoplasm of enterocytes by a protein called divalent metal transporter 1 (DMT-1). Once iron is within the enterocytes, it can be transported out into the blood by ferroportin, with some help from accessory proteins including ceruloplasmin and hephaestin. Once in the blood, the iron is bound by transferrin (eFig. 15.1). In contrast, if the body has sufficient iron stores, then the iron is not transported into the blood but remains within the cytoplasm of the enterocytes. Hepcidin blocks iron transport to the blood by degrading ferroportin. When the enterocyte eventually dies, the iron will be lost within the fecal stream, preventing iron overload (eFig. 15.2).
In healthy individuals, blood contains more transferrin protein than iron, with about 30% of the transferrin molecules saturated with iron. As blood iron levels increase, the excess transferrin protein serves as a reservoir that can quickly bind excess iron to prevent toxicity.
Iron Storage
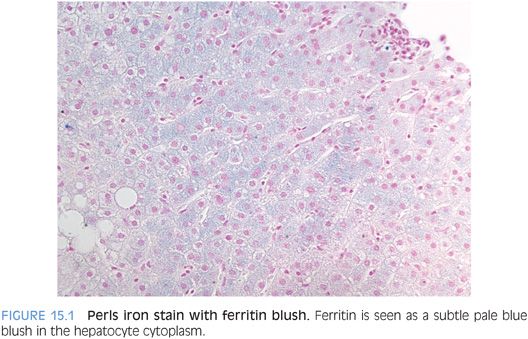
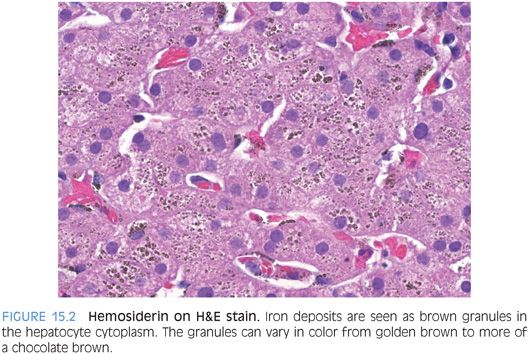
Within cells, principally in hepatocytes and macrophages, excess iron is incorporated into ferritin molecules for storage. Ferritin can hold up to 4,500 atoms of iron per ferritin protein complex. Ferritin is typically not visible on Perls Prussian blue stain, but occasionally ferritin is seen as a diffuse blue blush in the hepatocyte cytoplasm (Fig. 15.1). The iron in ferritin is readily available to meet physiologic needs. However, if ferritin levels are excessive over a sufficiently long period of time, hemosiderin deposits can develop. On hematoxylin and eosin (H&E) stain, hemosiderin is a granular, golden brown cytoplasmic deposit (Fig. 15.2). Hemosiderin is composed of iron along with degraded ferritin and small amounts of other proteins. In contrast to the iron stored as ferritin, the iron in hemosiderin deposits is not readily available for biologic needs.
In sum, two important iron reservoirs are used to keep blood iron levels at physiologically correct levels: (1) a short-term reservoir of iron stored within enterocytes and (2) a longer term reservoir of iron stored as ferritin, principally in hepatocytes and macrophages. If both reservoirs are unable to meet the demands for iron, then anemia develops; however, if iron control mechanisms are dysregulated (e.g., mutated) or if there is excess exogenous iron intake (e.g., transfusions), then iron overload can develop.
Iron Release from Stores in the Enterocytes, Liver, and Macrophages
Hepcidin is a major controller of iron metabolism1,2: It blocks the release of iron from hepatocytes, macrophages, and enterocytes into the blood (eFig. 15.3). When hepcidin levels are lowered, more iron is absorbed from the gut and more iron is released into the blood. Hepcidin (encoded by the gene HAMP) is also an acute phase reactant and is produced by hepatocytes3 and biliary epithelium.4 Because it is an acute phase reactant, hepcidin levels are elevated in many inflammatory and infectious conditions (Table 15.2). Hepcidin’s main physiologic role is to lower blood iron levels, which it does by blocking transfer of iron from enterocytes to the blood and by blocking the release of iron stores from the liver and macrophages into the blood. Hepcidin blocks iron transport by degrading ferroportin, the protein that exports iron out of cells into the blood.5 Ferroportin is the only known export protein for iron, and iron cannot be released from the enterocytes, macrophages, and hepatocytes into the blood stream without it.

MUTATIONS IN IRON-RELATED GENES
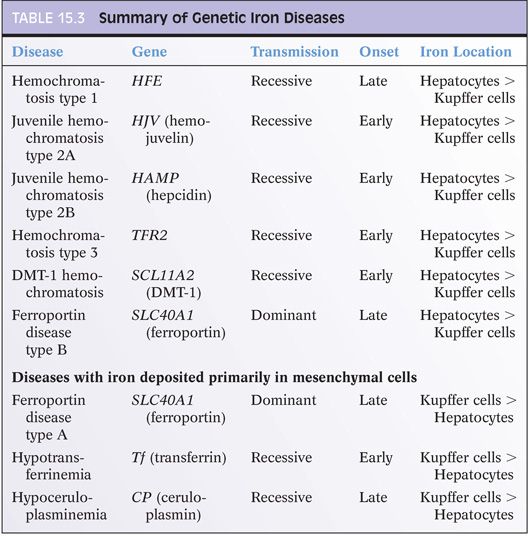
Hepcidin dysregulation plays a central role in essentially all known causes of hemochromatosis.1 In fact, mutations that lead to hemochromatosis all function by decreasing hepcidin production or impairing hepcidin function, including mutations in HFE, HAMP, HJV, and TFR2.6 Key features of these diseases are summarized in Table 15.3. The abnormally low levels of hepcidin gradually lead to excess iron absorption and eventually iron deposition in the liver and other organs. Interestingly, to date, most reported mutations lead to loss of hepcidin function. However, rare mutations have also been described that increase hepcidin function, with clinical manifestations of congenital refractory anemia.7 Increased expression of hepcidin (with subsequent development of anemia) has also been reported in a hepatic adenoma.8 Interestingly, hepatic adenomas occurring in individuals with type 1a glycogen storage disease are also associated with anemia that resolves after the adenoma is resected,9 implying hepcidin overexpression by the adenomas. In contrast, hepatocellular carcinomas tend to have suppressed levels of hepcidin expression.10
HFE Mutations
HFE mutations were first linked to hereditary hemochromatosis in 1996.11 Since that time, more than 37 mutations have been reported,12 but C282Y and H63D mutations are the most numerically and clinically important. Overall, C282Y mutations account for 80% to 90% of genetic hemochromatosis cases. Of the remaining cases, 60% are explained by H63D mutations.13,14 Other HFE mutations, such as S65C, have also been linked to iron accumulation,13 but the role of S65C in iron overloading is less clear.15 C282Y mutations are strongly associated with northern European genetic ancestry.12 H63D mutations also have a higher frequency in Caucasian populations but have a wider ethnic distribution.16,17
Individuals with C282Y mutations have a higher risk for iron accumulation than those with H63D mutations. Likewise, C282Y homozygotes have greater risk for iron accumulation than do C282Y heterozygotes. Nonetheless, there is great phenotypic variation, even for individuals who are homozygous for C282Y mutations. For example, one major population-based study from Australia followed 203 individuals who were homozygous for C282Y mutation for 12 years. During this time, 28% of men and 1% of women developed iron overload–related diseases.18 A similar study followed individuals who are compound heterozygous for C282Y/H63D mutation over the same 12-year interval and found that only 1 out of 82 men and none of 95 women developed iron-related disease.19 The striking phenotypic variation is likely related to gender, environmental factors, dietary factors, and other genetic polymorphisms.19
Clinical presentation is striking for its considerable variety. The classic presentation of cirrhosis, diabetes, and bronze skin that so many of us learned in medical school is now very uncommon because of earlier diagnosis. In many cases, individuals can present with vague findings of fatigue and bone and joint pain. Another common presentation is mild biochemical liver or iron blood work abnormalities identified while being evaluated for other conditions. Presentations later in the disease course can include varying combinations of endocrine dysfunctions—for example, adrenal insufficiency or diabetes mellitus—cirrhosis, heart failure, or joint disease.
Links between HFE Mutations and Other Chronic Diseases
For individuals with HFE mutations, other chronic liver diseases can affect iron overload risk. Likewise, HFE mutations have been linked to disease severity in other chronic liver diseases, such as chronic hepatitis C or fatty liver disease. Furthermore, some etiologies of cirrhosis, such as α1-antitrypsin deficiency or cryptogenic cirrhosis, can be enriched for HFE mutations and show marked iron accumulation.20
For the major causes of chronic liver diseases, such as chronic viral hepatitis and fatty liver disease, numerous studies have examined the relationship between disease severity and the presence of tissue iron accumulation and/or the presence of HFE mutations. Although the data is substantially mixed, the evidence supports an overall association between more severe disease and the presence of excess iron in the liver. However, the impact of liver iron accumulation on disease severity is typically modest, and there are many studies that could not identify an association. These negative studies highlight the difficulty of identifying a modest effect from within the very complex setting of clinical studies, where the challenge is to control for all of the factors that can influence iron status as well as fibrosis progression risk.
HEMOJUVELIN MUTATIONS (USUALLY CHILDREN/EARLY ONSET). Hemojuvelin-related iron disease is rare but is still the most common cause of juvenile hemochromatosis.21,22 In contrast to HFE-related disease, hemojuvelin disease typically presents with impotence or amenorrhea and not with liver or joint disease. Cardiomyopathy is also common at presentation.23 The hepatocytes can show marked iron overload. The disease typically runs a severe clinical course and can be rapidly progressive.2
The most common mutation (G320V) is found in 80% to 90% of cases of juvenile hemochromatosis. An I222N mutation has also been reported. In contrast to HFE mutations, the general population has a very low frequency of hemojuvelin mutations. For example, in a screen of 365 asymptomatic adults from Alabama, United States, only one I222N mutation and no G320V mutations were found.24
HEPCIDIN (USUALLY CHILDREN/EARLY ONSET). This rare form of genetic iron overload has marked hepatocellular iron overload and typically runs a severe clinical course. Hypogonadism and cardiac disease are prominent clinical manifestations.
TRANSFERRIN RECEPTOR GENE 2 (USUALLY ADULTS/LATE ONSET). This form of genetic iron overload is rare in Western populations and can have a variable clinical course, with some but not all individuals having marked hepatocellular iron accumulation. Also of note, polymorphisms in TFR2 are common in the general asymptomatic adult population, where they can lead to mild increases in blood iron levels without overt iron overload disease.25 Mutations have also been reported in other populations including Africa26 and Iran,27 but the overall epidemiology is unclear.
DMT-1 MUTATIONS (USUALLY OLDER CHILDREN). Mutations in the SLC11A2 gene are a very rare cause of genetic iron overload, and clinical and histologic data is quite limited.28 Children present with severe microcytic anemia. Iron accumulation is primarily in hepatocytes and can be severe,29 but biopsies can be negative for iron in very young children.
Nonhemochromatotic Iron Overload Disease (i.e., Mesenchymal Iron Accumulation)
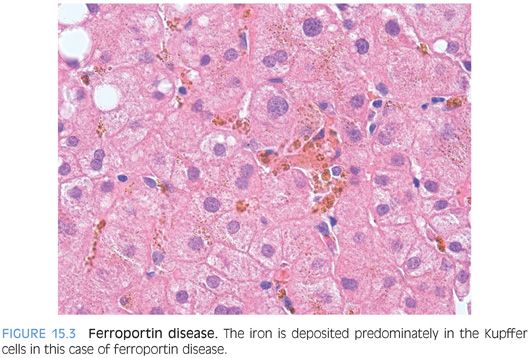
Ferroportin disease is a classic example of hereditary iron overload where the iron accumulation is predominately in Kupffer cells.30 Also of note, transferrin saturation levels in ferroportin disease do not become elevated until much later in the disease course, in contrast to other types of genetic hemochromatosis, which all have elevated transferrin saturation levels early in the disease course. Thus, high ferritin levels and low-to-normal transferrin saturation levels can be an important clue to the diagnosis in a patient with predominately Kupffer cell iron deposits. Ferroportin disease also stands out for its dominant inheritance pattern.2 Of note, there is substantial phenotypic variability and the disease is divided into two subtypes with different disease manifestations. Histologically, iron deposits in Kupffer cells predominate over that of hepatocytes in subtype A (Fig. 15.3), whereas hepatic iron is heavier than Kupffer cell iron in type B. Ferroportin disease can also have small sideronecrotic foci composed of iron-laden macrophages in small clumps within the lobules. A caveat is that the histologic data on ferroportin disease is somewhat limited and will only become clearer as larger case series are put together. Clinically, both types have milder disease than those with HFE mutations.
Neonatal Hemochromatosis
Despite the term hemochromatosis in neonatal hemochromatosis, neonatal hemochromatosis is fundamentally different than the other iron-related diseases previously discussed. Neonatal hemochromatosis is broadly classified as liver disease of the neonate accompanied by extrahepatic deposits of iron. The majority of neonatal hemochromatosis cases, but probably not all, result from an alloimmune gestational disease where maternal antibodies cross the placenta and attack the fetal liver in utero.31 The alloimmune target has not been clearly identified, but it is presumed to be an antigen on the hepatocyte cell surface. The newborn will typically have massive liver failure at birth or within the first few days of life. In many cases, there is late second-term or third-term fetal loss because the fetus did not survive the massive liver injury.
On biopsy of the liver (or on postmortem examination of the liver), the histology can range from massive liver necrosis with almost no residual hepatocytes to a severely damaged liver with regenerative nodules that can give the liver a cirrhotic appearance. The residual hepatocytes in these cases are often inflamed, cholestatic, and may show giant cell transformation. An iron stain will typically show hepatic iron accumulation with course granules of iron, qualitatively similar to that seen in adult hemochromatosis, whereas the Kupffer cells generally have little iron accumulation. As an important caveat, the histologic findings have not been described in many cases and it seems likely that there is more histologic variation than is currently reported. Serum α-fetoprotein (AFP) levels are also elevated in most cases, typically greater than 100,000 ng/mL, and they can be as high as 800,000 ng/mL (the healthy neonate typically has values less than 80,000 ng/mL).
Of note, extrahepatic iron deposits are helpful to confidently make this diagnosis based on histology. The best places to look are (in approximate order) acini of the pancreas, myocardium, thyroid, minor salivary glands, Brunner glands, pancreas islets, stomach, and chondrocytes.32 Lip biopsies can be helpful but often are not deep enough to get the glands needed to look for iron accumulation and thus are often not informative. The bone marrow and spleen often will not have increased iron. Also, if most of the hepatocytes have been destroyed, then there may not be much iron in the liver biopsy.
The prognosis is poor unless the disease is quickly recognized. Although data is limited, for those children who do survive, there does not appear to be any significant medical sequelae. Treatment of affected infants revolves around supportive care and removing the maternal antibody through plasmapheresis. If an infant is affected by this disease, there is about a 90% chance that subsequent pregnancies in the mother will be likewise affected, so this is an important disease to diagnose. Prevention for subsequent pregnancies is approached by treatment during gestation with intravenous immunoglobulin (IVIg) and is typically given from about the 18th week gestation until delivery.
More to Come
There remains a subset of hepatic iron overload cases that does not appear to have the mutations described earlier. For example, in a study from Brazil, one-third of cases with marked iron overload did not have the typical mutations discussed earlier.33 Similarly, iron overload in Africa is now recognized to be not exclusively diet-related, suggesting an unrecognized genetic component.34
DETECTION OF IRON IN THE LIVER
Iron Stains
The major histochemical stain used to detect iron in the liver is Perls Prussian blue, a stain named after Max Perls, a German pathologist. The basic chemistry of Perls Prussian blue is that iron in the ferric state will react with hydrochloric acid to form ferric ferrocyanide, an insoluble blue compound (Prussian blue) that is well visualized on light microscopy. The distribution and density of blue staining correlates well but not perfectly with tissue iron concentrations. The stain is not sensitive enough to detect very low levels of iron but is easy to perform and reproducible.
FERRITIN. Normally, no ferritin will be seen. However, in cases of elevated serum ferritin levels, ferritin may be seen as a light, diffuse, blue blush of the hepatocyte or Kupffer cell cytoplasm (see Fig. 15.1).
HEMOSIDERIN. Hemosiderin can be seen as brown granular cytoplasmic deposits on H&E stains (see Fig. 15.2) and as a bright blue granular staining on iron stain. Residual brown granular material is often seen on iron stain and represents lipofuscin in most cases (eFig. 15.4).
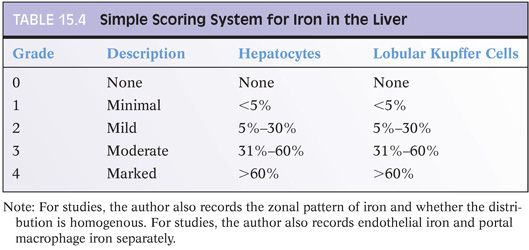
Iron Grading Systems
Many iron grading systems have been proposed over the years. They vary in their approach, but all attempt to provide semiquantitative data on the extent of iron accumulation. Some systems are based on the zonation of iron distribution, some on the lowest magnification that discernible granules can be seen, and some on the percentage of hepatocytes positive for iron. Is one system clearly the best? Probably not. The author personally uses a schema (Table 15.4) based on the percentage of hepatocytes positive for iron, similar to that described by LeSage et al.35 This simple-to-use classification system provides sufficient clinical information for patient care. But there are many reasonable alternatives to consider if you prefer a different approach. A modified Scheuer system (shown in Table 15.5) is also a very useful and popular system. If employed, separate numbers should be given for hepatocellular and the reticuloendothelial iron.