Infectious Colitis
General Comments
The indigenous colonic flora and an intact mucosal barrier represent vital components of the body’s defenses against invasion by pathogens. Disruption of these defenses facilitates bacterial translocation and contributes to disease severity. Colonic injury results from the presence of bacteria or their toxins. Pathogenic mechanisms of mucosal injury include bacterial adherence, invasion, and toxin production (see Chapter 6). The pathogens typically hijack the host cell cytoskeleton. Altering the cytoskeleton is crucial for mediating pathogen adherence, invasion, and intracellular locomotion, especially for E coli, Salmonella, and Shigella (196).
The risk of developing infectious colitis varies considerably throughout the world and depends on local conditions. Populations inhabiting developing countries often live in ramshackle housing without good sanitation. Enteric infections, particularly bacterial, are readily transmitted in this setting. In contrast, most inhabitants of industrialized countries live in a sanitary environment that generally discourages transmission of enteric pathogens. However, in industrialized countries, other practices facilitate bacterial transmission, including large-scale food production, distribution, and retailing practices, which create opportunities for widespread and extensive outbreaks of food-borne enteric infections (fast-food chains) (197). Infants in daycare centers, patients hospitalized in chronic care facilities, AIDS patients, travelers, and military personnel all have an increased risk of infection. Communication with the clinicians regarding patients’ travel, immune status, sexual practices, and food intake facilitates the detection of many infections.
Many gastrointestinal infections are acquired through the ingestion of contaminated food and water. The globalization of the food supply makes the effect of an accidental contamination or deliberate attack on it more widespread. If a bioterrorist attack occurs, pathologists could play a critical role in its identification. Table 13.11 lists factors that should trigger a suspicion of widespread food contamination.
Gastrointestinal changes vary from minimal changes to the classic pattern of acute self-limited colitis, or the production of nonspecific features or a necrotizing enterocolitis. Organisms that produce toxins tend to cause less severe morphologic changes than organisms that invade the mucosa (Fig. 13.95) (see Chapter 6). With the exception of enterohemorrhagic E. coli (EHEC), colitis-causing bacteria are invasive and include Shigella, Salmonella, Campylobacter, and Yersinia.
EHEC causes enterocyte damage via a secreted toxin. EHEC is the third most common bacterial pathogen isolated from stool samples from diarrheal patients, trailing only Salmonella and Campylobacter infections (198,199). Infective colitis also results from Yersinia enterocolitica (46% of cases), Campylobacter jejuni (20%), Salmonella (13%), and Shigella (9%) infections (199). Amebiasis and cytomegalovirus infections account for another 11% of cases (199). Of these infections, Salmonella and amebiasis mimic ulcerative colitis, whereas the other pathogens cause a focal colitis resembling Crohn colitis (199).
EHEC causes enterocyte damage via a secreted toxin. EHEC is the third most common bacterial pathogen isolated from stool samples from diarrheal patients, trailing only Salmonella and Campylobacter infections (198,199). Infective colitis also results from Yersinia enterocolitica (46% of cases), Campylobacter jejuni (20%), Salmonella (13%), and Shigella (9%) infections (199). Amebiasis and cytomegalovirus infections account for another 11% of cases (199). Of these infections, Salmonella and amebiasis mimic ulcerative colitis, whereas the other pathogens cause a focal colitis resembling Crohn colitis (199).
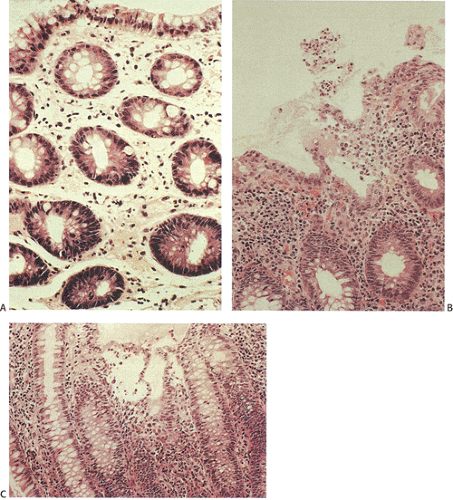 FIG. 13.95. Bacterial colitis. A: There is marked mucosal edema without obvious inflammation in a patient with a toxigenic Escherichia coli infection. B: Area of superficial ulceration and lamina propria inflammation. C: A small bulla has formed underlying the surface epithelium. B and C are from patients with two different types of invasive bacterial infections. |
TABLE 13.11 Factors Suggesting Widespread Microbial Food Contamination | |
|---|---|
|
TABLE 13.12 Location of Colonic Infections | |
|---|---|
|
The diagnosis of various forms of infectious colitis is based on a combination of clinical findings, histologic features, and response to therapy. The pathologic diagnosis of gastrointestinal infection depends on the recognition of several factors, including specific pathogens, specific tissue reactions, and specific cytopathic effects of the infection. The recognition of specific pathogens results from their localization to specific tissue sites, including the epithelial apical surfaces, the intestinal lumen, the lamina propria, the submucosa, the muscularis propria, or the myenteric plexus (Table 13.12). Special studies help identify specific pathogens. H&E stains often allow one to diagnose viral inclusions, but these may not always be obvious and the use of immunohistochemical reagents or even in situ hybridization reactions for a particular viral genetic sequence may prove informative. Special stains, particularly fungal stains, are often utilized to confirm the presence of infections, particularly in the setting of diffuse histiocytic infiltrates. Fungi may be highlighted with either Gomori or PAS stains. Microsporidia are highlighted by Gram stains or Giemsa stains, and acid-fast bacillus (AFB) stains are useful in identifying tuberculosis or Mycobacterium avium. Trichrome stains may help identify amoebae. Ultrastructural examination also aids in the identification of microsporidia and some protozoans. Cultures are often useful, as are stool analyses for the detection of specific toxins or the genes encoding those toxins. Increasing numbers of immunohistochemical stains are becoming available, as are probes for in situ reactions and other molecular methodologies.
TABLE 13.13 Histologic Features of Acute Self-limited Colitis | |
|---|---|
|
General Histologic Features of Bacterial Colitis (Acute Self-limited Colitis)
Histologic features of acute self-limited colitis (ASLC) include the features listed in Table 13.13 (Fig. 13.95). Table 13.14 lists the histologic characteristics at different times in the infection (200). The features of resolving colitis involve a shift from an acute inflammatory response to one that is more chronic in nature. The chronic changes of resolving infections may be more difficult to distinguish from other forms of colitis.
TABLE 13.14 Evolving Features of Acute Self-limited Colitis | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 13.15 Comparison of Acute Self-limited Colitis (ASLC) and Inflammatory Bowel Disease (IBD) | |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
In early infections the mucosa appears expanded by edema and patchy inflammation. Neutrophils are typically prominent in the lamina propria, often near dilated capillaries or alongside the crypts. Marginating neutrophils may be seen in the capillaries. Neutrophils also infiltrate the crypt epithelium, causing cryptitis. Crypt abscesses are not prominent at this stage of many infections. Neutrophils outnumber lymphocytes and plasma cells. The epithelium often appears ragged and degenerating, sometimes with syncytial tufts. The crypt lining may appear mucin depleted, flattened, or reactive. The crypts are arranged in parallel to one another and the upper halves may appear dilated. The lamina propria may contain fresh hemorrhage.
Since ASLC is clinically and endoscopically similar to active idiopathic IBD, pathologists are often asked to distinguish between these two entities (Table 13.15) (see also Chapter 11). An important histologic feature that distinguishes IBD from ASLC is the crypt architecture. When the architecture is normal, ASLC is likely (Fig. 13.5) and IBD is unlikely. Conversely, a distorted crypt architecture strongly correlates with IBD. The ability to correctly diagnose IBD is very high when the histologic findings include a distorted crypt architecture (branched or forked glands not caused by glands bending around lymphoid follicles), increased numbers of round cells and neutrophils in the lamina propria, a villous architecture, epithelioid granulomas, crypt atrophy (shortening and scarcity of glands), basal lymphoid aggregates, plasmacytosis, Paneth cell metaplasia, and basally located isolated giant cells. Villiform surface configurations usually occur in UC (Fig. 13.96). However, they are relatively uncommon and therefore have limited diagnostic value. One caveat with regard to the foregoing is that when ASLC occurs in populations with a high incidence of infectious disease, the patients may develop crypt distortion. This disorder is sometimes referred to as tropical colonopathy. Such patients may show effects of previous infection with organisms known to destroy crypt architecture, such as amoeba, superimposed on an acute bacterial infection.
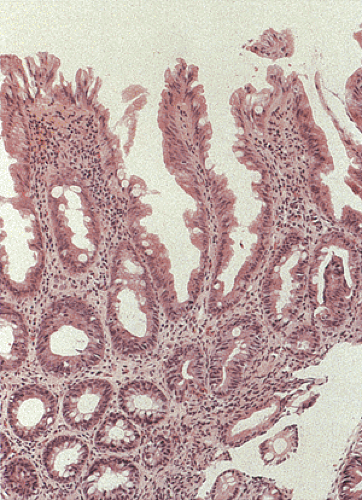 FIG. 13.96. Villiform transformation of the mucosa. |
The nature of the inflammation in the lamina propria also helps differentiate ASLC from acute-onset IBD. The presence of a pure acute neutrophilic inflammatory infiltrate suggests ASLC because this never occurs in IBD. However, a mixed acute and chronic infiltrate occurs in both diseases. Inflammatory changes in the basal lamina propria strongly suggest IBD. One reason for this is that the crypt bases are normally relatively acellular, and when plasma cells are increased in this area, they are easily detected. Both basal lymphoid hyperplasia and basal lymphoid aggregates highly discriminate for IBD because they are more common in IBD patients than in patients with ASLC. The basal lymphoplasmacytosis affects the lower 20% of the mucosa. Mononuclear cells, including lymphocytes and plasma cells, may increase in ASLC but they are usually not present to the degree found in IBD unless the IBD patients have been treated.
The resolving form of an acute self-limited colitis is particularly difficult to distinguish from IBD. Inflammation limited to the superficial one half or two thirds of the lamina propria occurs more frequently in patients with ASLC, but this feature is not always present. The inflammation may form bandlike inflammatory infiltrates preferentially lying in the upper or midzonal parts of the mucosa and sparing the lower mucosa. The inflammation occupies the lamina propria rather than the crypt epithelium as in idiopathic IBD. Edema and hemorrhage, other hallmarks of infectious colitis, are absent or inconspicuous in idiopathic IBD. Focal inflammation occurs more commonly in patients with ASLC than in patients with UC.
TABLE 13.16 Granulomas in Bacterial Colitis | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
Some infections produce epithelioid granulomas similar to those seen in CD. Others elicit microgranulomas or histiocytic collections lacking giant cells (Table 13.16). Inconspicuous microgranulomas may be seen in ASLC, particularly with Campylobacter and Salmonella infections, although these are rare. Granulomas are more frequent in Yersinia enterocolitica and Mycobacterium tuberculosis infections.
Specific Bacterial Infections
Escherichia coli
As indicated in Chapter 6, humans develop several types of E. coli infection. Toxigenic strains tend to cause minimal injury (Fig. 13.97). EHEC infections are discussed here; the remaining E. coli infections are covered in Chapter 6.
Enterohemorrhagic colitis results from infections due to Shiga toxin–producing E. coli. E. coli 0157:H7 is one strain of Shiga toxin–producing E. coli (201,202,203) and the one most commonly associated with HUS in the United States. Other strains cause HUS in other parts of the world. Two similar but distinct bacterial cytotoxins (Shiga-like toxins I and II) contribute to the pathogenicity of enterohemorrhagic E. coli infections. These toxins have two units that are almost identical in structure to Shiga toxin and cholera toxin. Both toxins interact with the same membrane receptor (202). A unique plasmid-encoded fimbrial adhesin facilitates adherence of E. coli 0157:H7 to the intestinal mucosa (203). The organism targets Peyer patches, where it creates an attaching/effacing lesion (204). As a result, EHEC does not invade the epithelium but rather adheres to the luminal surface, where it elaborates a toxin. The absorbed toxins interfere with protein synthesis, causing epithelial and endothelial damage (205). These toxins damage the vascular endothelium of the kidneys and intestines and mediate bacillary dysentery, hemorrhagic colitis, HUS, and thrombotic thrombocytic purpura (TTP) in infected patients (206). The damaged endothelium fails to secrete anticoagulants, initiating microvascular thrombosis. Because of the underlying endothelial damage, the tissues often exhibit morphologic changes indistinguishable from ischemic colitis. Not all EHEC strains carry a risk for the development of HUS. HUS develops in 5% to 8% of patients with EHEC infection. High white blood cell counts, elevated C-reactive protein, and fever early in the course of the disease may be indicators of risk for development of HUS (207,208).
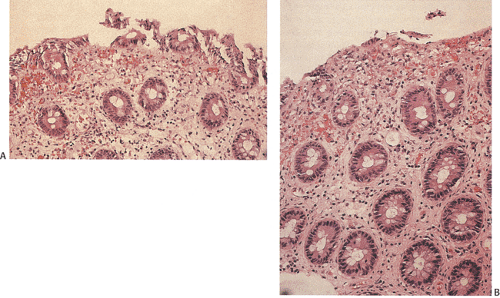 FIG. 13.97. Toxigenic Escherichia coli infection. A and B are from two different areas of the colon. A shows less damage than B, which is almost completely denuded. Both figures demonstrate superficial edema with extravasation of red blood cells. The biopsy is from a 17-year-old boy who ate a hamburger at a picnic and whose stool was positive for the E. coli toxin. |
The mean annual rates of E. coli 0157:H7 infection range from 2 per 100,000 to 12.1 per 100,000 in two different parts of the world (209). In the United States, E. coli 0157:H7 infections are widespread, as shown by the number of outbreaks and sporadic cases, severe illnesses, and deaths. In one study, E. coli 0157:H7 was the fourth most common bacterial
stool pathogen (210). Another study by the Centers for Disease Control and Prevention showed that 8% of routine cultures were positive for this organism, with an infection rate higher than that seen for Shigella (211). The infection occurs nationwide, although infection rates in the South are lower than those in the rest of the country. Patients range in age from 1 to 80 years, with a median age of 14 years. Populations most susceptible to the infection are the very young and the very old.
stool pathogen (210). Another study by the Centers for Disease Control and Prevention showed that 8% of routine cultures were positive for this organism, with an infection rate higher than that seen for Shigella (211). The infection occurs nationwide, although infection rates in the South are lower than those in the rest of the country. Patients range in age from 1 to 80 years, with a median age of 14 years. Populations most susceptible to the infection are the very young and the very old.
The incidence of the infection is highest in the summer, with most cases occurring between May and September (212). E. coli 0157:H7 outbreaks usually occur in communities, nursing homes, daycare centers, kindergartens, and children’s wading pools (213). E. coli 0157:H7 can survive in drinking water and associate with water-borne disease outbreaks (214). The infectious dose of EHEC has been estimated to be fewer than 100 organisms. Shiga toxin–producing E. coli also associates with the consumption of hamburgers, other beef products, unpasteurized milk, cheese, pork, poultry, lamb, vegetables, and fruits (215,216). Ground beef represents an especially important vehicle of disease transmission (217). The E. coli 0157:H7 in ground beef is more sensitive to heat than other bacteria, but it survives for months at -20°C (218). For this reason, proper cooking is the preventive measure of choice. Person-to-person transmission is also possible, as is transmission by environmental contamination (219).
Currently, many laboratories do not routinely perform surveillance cultures for E. coli 0157:H7 unless the stool specimen is bloody or a specific request is made for it. Therefore, the infection often goes undetected (220). Screening for E. coli 0157:H7 is performed with the use of sorbitol MacConkey agar (218).
The mean incubation period is approximately 3 to 4 days and the illness usually lasts for 2 to 9 days. Asymptomatic infections are common, as is a self-limited, nonbloody, afebrile diarrhea. Other patients develop ileocolitis. Young children, the elderly, and immunosuppressed individuals are more susceptible to HUS and TTP. Antimicrobial treatment does not influence symptom duration, nor does it alter the risk of developing HUS or TTP. Patients with colitis develop bloody diarrhea, severe crampy abdominal pain, and low-grade fever. Fecal leukocytes are absent. Complications include thrombocytopenic purpura, end-stage renal disease, and neurologic damage, including strokes (221).
Endoscopy demonstrates the presence of a normal mucosa with oozing of blood. Other changes include mucosal edema and congestion, erosions, ulcers, erythema, a friable mucosa, and the presence of exudates or pseudomembranes. The submucosa appears swollen. The edema may be so marked that obstruction develops, requiring surgical resection. Resection is also used to control bleeding. The lesions often localize to the right colon but pancolitis may develop, particularly in children.
The histologic features demonstrate overlapping ischemic and infectious patterns due to the presence of both epithelial and endothelial injury (Fig. 13.98). Features associated with the ischemic damage include focal marked submucosal edema, hemorrhage, pseudomembranes (221), bland mucosal necrosis, ulceration associated with fibrin thrombi in the mucosal and submucosal capillaries, and abundant intramural fibrin deposits. The ischemia leads to extensive areas of mucosal necrosis, leaving ghosts of crypts with underlying neutrophilic infiltrates. The necrosis may extend to the submucosa or transmurally with an overlying serosal exudate. The intervening mucosa remains minimally affected with mild mucus depletion and occasional neutrophils and capillary platelet thrombi. The intervening mucosa may also exhibit epithelial dropout, fibrin deposition, and adjacent hemorrhage.
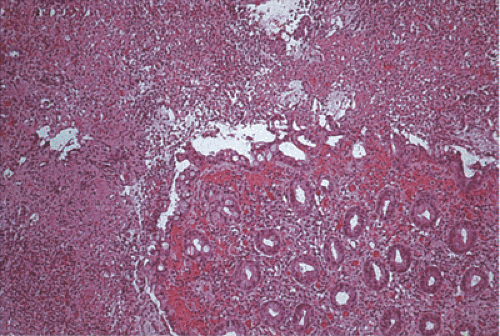 FIG. 13.98. Escherichia coli 0157:H7. This patient had a hemorrhagic colitis with pseudomembrane formation. |
Histologic features associated with an infectious histologic pattern include a prominent neutrophilic infiltrate within the crypts, lamina propria, and adherent pseudomembranes. The neutrophilic infiltrate may be mild and patchy, or it may infiltrate the crypt epithelium, producing cryptitis or incipient crypt abscesses. Cryptitis occurs more commonly than crypt abscesses, and in all cases the neutrophilic infiltrate is focally accentuated rather than uniform in appearance, and may even appear as solitary inflammatory foci. Patients often exhibit extreme submucosal edema with hemorrhage and fibrin exudation. The focal nature of the neutrophilic infiltrate, along with the predominance of cryptitis over well-formed crypt abscesses, resembles the histologic changes seen in Campylobacter, Salmonella, and early amebic infections. Its focal distribution and the presence of fibrin thrombi distinguish the infection from acute UC. The changes may resemble those present in C. difficile colitis. A disproportionate amount of lamina propria hemorrhage is often present when compared to the degree of inflammation. Glandular distortion, architectural abnormalities, granulomas, and giant cells are usually absent unless there is underlying pre-existing IBD (222). Because the disease is patchy, biopsies may appear normal or only exhibit a focal, mild, nonspecific increase in lamina propria lymphocytes or plasma cells. The histology does not correlate with either disease duration or the
course of the illness. Advanced lesions display regenerative mucosal changes and heavy submucosal plasmacytic infiltrates.
course of the illness. Advanced lesions display regenerative mucosal changes and heavy submucosal plasmacytic infiltrates.
The differential diagnosis includes C. difficile colitis and ischemic colitis, which share overlapping features with the E. coli infection. The C. difficile antigen test may be very helpful in excluding C. difficile infections. Culture and serotyping are the mainstays of the diagnosis. Immunohistochemical stains and molecular assays for EHEC exist, but they are not widely available.
Campylobacter Infections
Campylobacter infections affect both the large and small intestines. Occasionally, C. jejuni is responsible for an acute focal colitis (223). Severe cases may present as an acute toxic ulcerative colitis with pancolitis, toxic megacolon, and colonic perforation. These infections are discussed further in Chapter 6.
Salmonella Infections
As discussed in Chapter 6, Salmonella infections cause five clinical syndromes. In this chapter, we will focus on Salmonella infections that present as colitis. Salmonella gastroenteritis results from infection by Salmonella enteritidis, Salmonella typhimurium, Salmonella argona, Salmonella javiana, Salmonella poona, Salmonella oranienburg and Salmonella newport. It varies from a mild to a severe infection, occasionally associated with bacteremia or bacteriuria. Salmonella gastroenteritis is acquired by ingestion of infected foods or drinks accounting for up to 80% of food poisonings. Most outbreaks occur through June and July and associate with consumption of contaminated fish, shellfish, cheese, and poultry (224,225,226,227). There is also a high prevalence of Salmonella in livestock. The practice of feeding subtherapeutic amounts of antibiotics to improve animal growth has resulted in the emergence of multiple antibiotic-resistant Salmonella strains in the animals and an increased incidence of antimicrobial-resistant organisms causing serious human disease. Food handlers who may become human carriers are occasionally implicated in spread of the infection.
The northeastern portion of the United States and parts of Europe has experienced a marked increase in food poisoning due to S. enteritidis. S. enteritidis is also increasing in South America and Africa (228). The increase relates to increased consumption of infected eggs and poultry. Approximately 400 cases of salmonellosis are reported annually in the United States, with fatality rates ranging from 1.3% to 8.4% (229).
Salmonellosis usually causes a mild self-limited disease that develops 8 to 48 hours following organism ingestion. Signs and symptoms of Salmonella gastroenteritis vary widely, but most patients present with nausea, vomiting, abdominal cramps, fever, pain, and diarrhea, which sometimes becomes bloody. Symptoms last for 3 to 12 days. Salmonella infections in the elderly have a poorer prognosis than when they occur in younger individuals. This is because the patients are more likely to become septic. Rarely, life-threatening, massive lower GI bleeding occurs. Bleeding usually arises in the small intestine but large intestinal bleeding also occurs (230). Patients not only develop GI symptoms, but also may develop erythema nodosum, reactive arthropathy, and rectal prolapse, particularly those with massive diarrhea. A pseudoappendicitis or ileocecitis may occur and may be complicated by perforation or toxic megacolon. Infections are particularly virulent in individuals with sickle cell disease or those with HIV infections. Diffuse or patchy mucosal hyperemia, friability, and inflammation may be seen at the time of colonoscopy.
Salmonella bacteria are intracellular parasites that enter the host by penetrating intestinal epithelial barriers, usually in the area of Peyer patches in the M cells. Once they are in close contact with the epithelium, the bacteria induce degeneration of the enterocyte microvilli (231), which is followed by profound membrane ruffling in the area of the bacterial host cell contact. Profuse micropinocytosis leads to bacterial internalization (232). Salmonella entry into the epithelium requires several chromosomal genes (inv/spa) clustered in a pathogenicity island termed SPI1 (salmonella pathogenicity island 1) (233). Additional pathogenicity factors are summarized in the Armed Forces Institute of Pathology (AFIP) fascicle (234). Bacteria enter membrane-bound vacuoles (phagosomes) inside epithelial cells and macrophages where they replicate (235). Replication also occurs in macrophages of the lymphoid follicles leading to bacteremia, reinfection of additional macrophages, and seeding of distant sites. As a result, the lymphoid follicles become hyperplastic, swollen, congested, and ulcerated, often resulting in typical longitudinally oriented ulcers and areas of hemorrhage. Edema, fibrinous exudation, and vascular thrombosis precede the ulceration. Aphthous ulcers sometimes develop.
Histologically mild cases show nonspecific patchy changes consisting of edema, congestion, and focal inflammation (Fig. 13.99). More severe cases show crypt abscesses with prominent neutrophilic infiltrates in degenerating crypts. The neutrophilic infiltration is more intense in the lamina propria than in the glands. Areas of hemorrhage and ulceration are also present. Crypt abscesses may be present in the nonulcerated areas. There may also be extensive areas of mucosal necrosis and hemorrhage with punched-out mucosal ulcers or erosions with elevated borders, crypt abscesses in the nonulcerated areas, and extensive mucosal and submucosal necrosis and hemorrhage (Fig. 13.100). These changes differ from IBD by the relative scarcity of chronic inflammatory cells. Microthrombi fill small mucosal and submucosal venules, creating a picture resembling that seen in acute ischemia. In severe cases, giant cells may populate the inflamed tissues, which, if transmural inflammation is present, might suggest the presence of CD. However, the giant cells do not form compact granulomas, as seen in CD. Occasional patients will show mild crypt distortion and branching, especially in those with persistent diarrhea.
There is sometimes a concurrence between salmonellosis and IBD (236), creating diagnostic and treatment dilemmas.
Rectal biopsies show the active inflammation typical of UC (237). The immunosuppression used to treat the IBD may predispose to the infection.
Rectal biopsies show the active inflammation typical of UC (237). The immunosuppression used to treat the IBD may predispose to the infection.
 FIG. 13.99. Salmonella colitis. A: The mucosa is inflamed and there is glandular destruction. There are apoptoses in one of the crypt bases mimicking drug injury and graft versus host disease. B: The base of several crypts show increased mitotic activity. The surrounding lamina propria appears edematous and the vessels are congested. |
Shigellosis
Shigella is a nonmotile, Gram-negative bacillus that is among the more virulent human enteropathogens. Shigella infections are an important cause of morbidity and mortality in developing countries, particularly in tropical areas. It causes approximately 10,000 cases of gastroenteritis each year in the United States (238). Shigella infections are more common in AIDS patients. They have a more virulent course and the infection is difficult to treat so that recurrences are common. Outbreaks of shigellosis among men who have sex with men result from direct or indirect oral–anal contact and are usually cause by Shigella flexneri. However, there was an outbreak of Shigella sonnei in California in 2000–2001 among men having sex with men (239). Person-to-person transmission and ingestion of contaminated food and water also cause the disease (240,241). Shigella infections associate with poor hygiene and overcrowding, and are a significant problem among children in nurseries and mental hospitals. Several Shigella species (dysenteriae, flexneri, boydii, and sonnei) cause colitis, with diminishing severity from the first organism to the last (242). S. sonnei, which produces a mild colitis, is the most common cause of bacillary dysentery in the Western world. Shigellosis results from release of bacterial toxins into the bowel lumen, as well as from direct bacterial invasion of the colonic mucosa (243). Shigella produces a potent toxin that inhibits protein synthesis and has cytotoxic, neurotoxic, and enterotoxic effects (244).
Children are the main victims of Shigella infections. The two major clinical presentations include watery diarrhea and dysenteric syndromes. The watery diarrhea has a short duration before hospitalization. The dysenteric form presents with bloody mucoid stools (27%), intense crampy abdominal pain (94%), diarrhea (98%), fever (87%), and nausea and vomiting (78%). It has a longer illness duration prior to hospitalization (245). Septicemia, hyponatremia, and hypoglycemia are common (246). The small volumes of dysenteric stool contain blood and pus. Patients with longer symptom duration may develop relative vascular insufficiency, activated lymphocytes, eosinophilic and mast cell degranulation, and antibody-mediated damage.
The severity of Shigella infections depends on many factors, including previous exposure to the organism and the infecting dose. The infections are usually self-limited, but dysentery can be a life-threatening illness in infants, the elderly, or malnourished individuals. The usual incubation
period ranges from 1 to 3 days. Watery diarrhea, with or without vomiting, is the initial symptom. After 24 hours, the stool becomes mucoid and grossly bloody. Low abdominal pain is common. Severe disease clinically mimics toxic megacolon, with hemorrhage, paralytic ileus, and perforation. Anal or perianal disease, including fissures, fistulae, hemorrhoids, or prolapse (245), complicates severe diarrhea. Serious complications are rare, especially in infections by S. flexneri and S. sonnei. However, patients with Shigella dysenteriae infection may develop bacteremia, sepsis, paralytic ileus, toxic megacolon, disseminated intravascular coagulation, and renal cortical necrosis severe enough to require hemodialysis (247). Shigellosis also causes hemolytic uremic syndrome, a condition associated with a high mortality (248). Circulating endotoxins cause the coagulopathy, renal microangiopathy, and hemolytic anemia. Other complications include toxic megacolon, intestinal perforation, sepsis, encephalopathy, pneumonia, conjunctivitis, and arthritis (249).
period ranges from 1 to 3 days. Watery diarrhea, with or without vomiting, is the initial symptom. After 24 hours, the stool becomes mucoid and grossly bloody. Low abdominal pain is common. Severe disease clinically mimics toxic megacolon, with hemorrhage, paralytic ileus, and perforation. Anal or perianal disease, including fissures, fistulae, hemorrhoids, or prolapse (245), complicates severe diarrhea. Serious complications are rare, especially in infections by S. flexneri and S. sonnei. However, patients with Shigella dysenteriae infection may develop bacteremia, sepsis, paralytic ileus, toxic megacolon, disseminated intravascular coagulation, and renal cortical necrosis severe enough to require hemodialysis (247). Shigellosis also causes hemolytic uremic syndrome, a condition associated with a high mortality (248). Circulating endotoxins cause the coagulopathy, renal microangiopathy, and hemolytic anemia. Other complications include toxic megacolon, intestinal perforation, sepsis, encephalopathy, pneumonia, conjunctivitis, and arthritis (249).
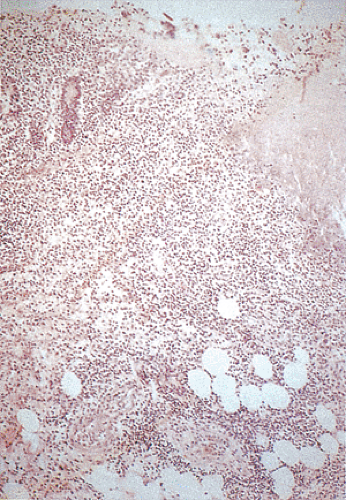 FIG. 13.100. Typhoid ulcer extending into the submucosa. The mucosa is inflamed and necrotic. |
Shigella organisms pass from the mouth to colonize the colon. Small intestinal infections do not occur unless the patient has a motility disturbance. Once they reach the colon, the bacteria penetrate the intestinal mucous layer and invade the epithelium. Multiple genes control epithelial entry (250). The preferential site of entry is through the dome of the lymphoid follicles. An adhesive or invasive phenotype is required for efficient colonization of the M cells of the follicle-associated epithelium. The invasive phenotype causes major inflammation-mediated tissue destruction, whereas the adhesive phenotype causes alterations in the M cells, which become stretched over large pockets containing aggregates of mononuclear cells. The M cells progressively occupy larger surface areas of the follicle-associated epithelium, causing the enterocytes to come off the epithelial surface (251). The organisms that invade the epithelium reorganize the cell cytoskeleton, a process that requires Shigella Ipa proteins. Once inside the cells, bacteria lie within membrane-bound vesicles. Here they multiply, causing mucus secretion and goblet cell depletion and inducing luminal neutrophilic infiltrates. The neutrophils loosen the intercellular barriers, further facilitating Shigella invasion (252).
Shigella bacteria direct their own uptake into the colonic mucosa through membrane ruffling and macropinocytosis in a manner similar to Salmonella uptake (253). After engulfment, the bacteria are surrounded by a membrane-bound vacuole within the host cell. Shigella rapidly lyses the surrounding vacuole and it is released into the cytosol where it grows and divides (251). Once it escapes from the vacuole it is quickly coated with filamentous actin and ultimately forms an actin tail at one pole of the bacterium (254). This actin polymerization propels the bacterium through the cell cytoplasm (255) and when the pathogen reaches the plasma membrane, it forms a long protrusion into the neighboring cell, which subsequently internalizes the microbe (256). The bacterium again breaks out of the vacuole starting a new cycle of infection in a new host cell (257). The cycle of intracellular and intercellular infection allows colonization of large epithelial surfaces while the bacterium is protected from immune host surveillance mechanisms. Bacilli penetrate the intestinal epithelium and pass through the mucosa in a period of several hours (251). The organism induces apoptosis in macrophages, which in agony release mature IL-1β. The IL-1β attracts neutrophils to the site of infection resulting in the massive colonic inflammation characteristic of the disease (258).
The Shigella cytotoxin rapidly reduces epithelial protein synthesis. An endotoxin damages mitochondria, leading to further destruction of cellular organelles, cell death, extrusion, and the formation of microulcers in the surface epithelium. Inflammation results in focal abscess and ulcer formation and, when severe enough, ileus, toxic megacolon, gross hemorrhage, and perforation. Endotoxin absorption in the lumen also leads to thrombosis, hemorrhage, and vascular insufficiency, causing further crypt damage.
Endoscopically, the mucosa appears edematous, friable, hyperemic, and ulcerated. All patients have inflammation in the rectosigmoid area. In most patients, the lesions are continuous and diffuse with the intensity of inflammation decreasing as one moves proximally. In severe cases there is a pancolitis with backwash ileitis, which can extend as far as 50 cm from the ileocecal valve (246). There may also be coexisting appendicitis. Serpiginous ulcers develop on the free edge of the mucosal folds oriented transversely to the long axis.
The intervening mucosa appears granular and hemorrhagic. Aphthous erosions may be present (Fig. 13.101). Other causes of aphthous ulcers are listed in Table 13.17. In fatal infections a gray mucopurulent exudate covers the mucosa. Because the findings resemble those seen in Campylobacter and Salmonella infections and IBD, definitive diagnosis requires stool culture.
The intervening mucosa appears granular and hemorrhagic. Aphthous erosions may be present (Fig. 13.101). Other causes of aphthous ulcers are listed in Table 13.17. In fatal infections a gray mucopurulent exudate covers the mucosa. Because the findings resemble those seen in Campylobacter and Salmonella infections and IBD, definitive diagnosis requires stool culture.
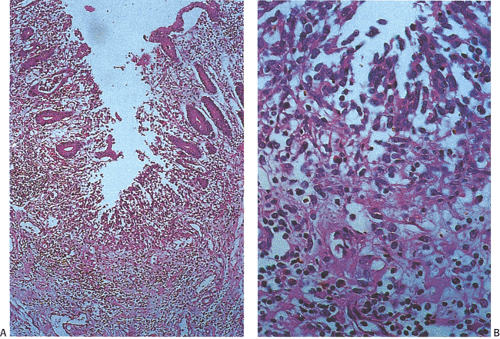 FIG. 13.101. Shigellosis. A: Mucosal ulceration and transmural inflammation. B: Higher power magnification of fibrinonecrotic debris. |
Histologic changes in Shigella infections include patchy hyperemia and edema with mucosal friability or ragged ulcerations, sometimes associated with pseudomembrane formation. Biopsies of early-stage disease show a surface epithelium that is reduced in height and infiltrated by neutrophils. The epithelial cells then detach from an edematous lamina propria forming microulcers. Neutrophils frequently lie below the microulcers. Detached epithelial cells, neutrophils, and red blood cells form a layer of purulent exudate on the surface. Microulcers and erosions are present in 85% of biopsy samples taken during the acute stage of the disease (246). Small aphthous ulcers also form. These always arise over the lymphoid follicles because the follicles with their M cells are the portal of entry (Fig. 13.101). Other changes consist of acute inflammation with neutrophilic emigration, superficial crypt abscess formation, early goblet cell depletion, edema, erythema, and mild plasma cell infiltrates (224). Shigella bacteria are numerous in the epithelial cells and in the lamina propria of early lesions. Dilated vessels accompany marked edema. As the lesions become more advanced, epithelial necrosis occurs and purulent exudates cover the damaged mucosa. Frank ulceration develops, disturbing the crypt architecture and causing pseudopolyp formation. The changes may remain confined to the mucosa, although they can extend into the submucosa. Patients may show mild crypt distortion and branching. Cellular regeneration progresses from the base of the crypts toward the mucosal surface. Even in the case of extensive mucosal damage, recovery is usually very rapid.
TABLE 13.17 Disorders Associated with Aphthous Ulcers | ||||||||
|---|---|---|---|---|---|---|---|---|
|
In autopsy cases, chronic ulcers of variable size and depth are present in 100% of cases. The ulcers may be superficial, involving only the upper quarter of the mucosa, but they may extend down to the muscularis mucosae. Some patients have wide areas of mucosal denudation and pseudomembranes. Crypt abscesses may be present along with architectural distortion and thrombosis of small vessels in the mucosa or submucosa. Colitis cystica profunda develops in about a quarter of the cases (246).
Convalescent stage biopsies show residual small superficial ulcers and marked inflammation of the lamina propria. The intensity of the lymphocytic and plasmocytic infiltrate is greater than that of the neutrophils.
Clostridium difficile
C. difficile is a well-known cause of epidemics and is the most common cause of nosocomial diarrhea. There are four groups of risk factors for the development of the disease: Patient risks, treatment risks, environmental risks, and the C. difficile strain (Table 13.18) (259,260). Infants, children, and adults may acquire C. difficile infections when they are hospitalized (261,262). The patients often receive antibiotic therapy in a setting where environmental contamination with C. difficile spores is commonplace (263). In one study, 21% of patients acquired C. difficile during their hospitalizations. Of these, 63% remained asymptomatic; 37% developed diarrhea. Among patients in the community who are treated with oral antimicrobial agents, only 1 to 3 individuals per 100,000 develop C. difficile colitis compared with as many as 1 per 100 hospitalized patients treated with similar drugs (263). Immunodeficient patients are particularly susceptible to developing C. difficile infections (264).
TABLE 13.18 Risk Factors for Acquiring Clostridium difficile Infections | |
|---|---|
|
C. difficile spores are heat resistant and may persist in the environment for months. The organism may spread person to person by hands, via fomites, or by general contamination (265). The organism can be cultured from hospital floors, toilets, bedding, and furniture, especially in areas where patients with diarrhea from C. difficile infection have been recently treated. Features that determine whether or not a patient develops a C. difficile infection include the nature of the fecal flora, the size of the C. difficile population, production of the requisite cytotoxins, and the presence of other organisms that affect toxin expression or activity. Host risk factors include advanced age, severe underlying illness, and prolonged hospital stay. Recent studies suggest that immunologic susceptibility has a role in C. difficile infection. The presence of an IgG antibody against toxin A protects against the clinical expression of a C. difficile infection and against relapse (266,267).
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


