Inflammatory bowel disease (IBD) is an immune-mediated disease and involves a complex interplay of host genetics and environmental influences. Recent advances in the field, including data from genome-wide association studies and microbiome analysis, have started to unravel the complex interaction between host genetics and environmental influences in the pathogenesis of IBD. A drawback of current clinical trials is inadequate or lack of immune phenotyping of patients. However, recent advances in high-throughput technologies provide an opportunity to monitor the dynamic and complex immune system, which may to lead to a more personalized treatment approach in IBD.
Key points
- •
Most of the recent advances in inflammatory bowel disease (IBD) have resulted from studies of mucosal immunity in the normal and inflamed intestine.
- •
Both murine models of IBD and human studies have shown dysfunction of the epithelial barrier, innate immune cells, and adaptive T cells in the pathogenesis of IBD.
- •
The insight gained from the study of the aberrant immune system in IBD has led to the identification of molecular targets in the immune system for the design of drugs, some of which are already being used in clinical practice with many others in various phases of development.
- •
Despite the increased knowledge gained from animal and human studies, many aspects of mucosal immunity remain unclear in patients with IBD.
- •
Recently, significant progress has been made in high-throughput technologies like genomic sequencing and mass cytometry that provide multiparametric data which can be used to not just define the various immune cells states but also assess how these interact with each other in a variety of conditions.
Introduction
Inflammatory bowel disease (IBD), specifically Crohn disease (CD) and ulcerative colitis (UC), are autoimmune diseases whose incidence and prevalence are increasing worldwide. Over the last few decades, substantial progress has been made in understanding the pathophysiology of IBD, which has been translated into newer, more effective therapies (biologics) that have reduced flares, brought more patients into remission, and improved the quality of life of patients with IBD. IBD is considered to be an immune-mediated disease that involves a complex pattern/interplay of host genetics and environmental influences. Our knowledge of the immune system and its homeostatic imbalance is derived from mouse models of colitis and human studies involving clinical and laboratory experiments.
The immune system evolved in multicellular organisms/metazoans as a defense mechanism against pathogens like bacteria, protozoa, parasites, and fungi. The human immune system can be broadly categorized into innate and adaptive based on the differences in timing of the response and specificity. The immune system comes in contact with a foreign challenge, which could be food, commensal flora, microbial pathogens, and xenobiotics at different sites like the skin, mucous membrane of lungs, gastrointestinal tract, and so forth. The human gastrointestinal tract, with a total surface area roughly equal to that of a tennis court (400 m 2 ), serves as the largest area of interface with the external environment. The gut mucosal immune system, which interacts with this large antigenic load, thus, has the most varied immune cells in the body. In a disease-free host, there is a fine balance between a protective and deleterious response of the immune system, which becomes perturbed in patients with IBD. To understand these perturbations in IBD that produce a disease state, it is necessary to first understand how the intestinal immune system works. In this review, the authors divide their article into subsections of innate and adaptive immunity and link it with the currently identified abnormalities in these pathways in IBD. In addition, the authors have summarized in Table 1 the current and emerging therapies for IBD that target specific molecules in the immune system.
| Biologic Target | Antibody/Drug | Mechanism of Action | CD, UC, or Both |
|---|---|---|---|
| CCR9 | CCX282-B | Inhibition of CCR9 | CD |
| CCX 025 | Inhibition of CCR9 | CD | |
| IL-21 | PF 05230900 | IL-21 receptor antagonist | CD |
| IL-13 | QAX576 | IL-13 antagonist | CD |
| Anrukinzumab | IL-13 antagonist | UC | |
| Tralokinumab | IL-13 antagonist | UC | |
| IL-17 | Vidofludimus | Inhibitor of IL-17 A and IL-17F | Both |
| IL-12/23 | Ustekinumab | Blockade of IL-12/23 | CD |
| IL-18 | GSK1070806 | Blockade of soluble IL-18 | CD |
| IL-6 and IL-6R | Tocilizumab | Inhibitor of IL -6 | CD |
| PF04236921 | Inhibitor of IL -6 | CD | |
| IP-10 | MDX 1100 | Blockade of interferon-γ inducible protein (IP-10 or CXCL10) | UC |
| IRAK4/TRAF6/MyD88 | RDP58 | Disrupts IRAK4/TRAF6/MyD88 signaling and reduces production of proinflammatory cytokines | Both |
| JAK3 | Tofacitinib | Inhibition of JAK3 | Both |
| MAdCAM-1 | PF-547659 | Blocks MAdCAM-1 | Both |
| NF-κB | HE3286 | Synthetic steroid that modulates NF-κB activity | UC |
| NKG2D | NN8555 | Anti-NKG2D receptor monoclonal antibody | CD |
| PKC | AEB071/Sotrastaurin | PKC inhibitor | UC |
| T Cell | Laquinimod | Reduces IL-17 level and interferes with migration of T cells | CD |
| TLR | DIMS0150 | Blockade of Toll-like receptor | UC |
| BL-7040 | Blockade of Toll-like receptor | UC | |
| TNF-α | Infliximab | Neutralization of TNF-α | Both |
| Adalimumab | Neutralization of TNF-α | Both | |
| Certolizumab pegol | Neutralization of TNF-α | CD | |
| Golimumab | Neutralization of TNF-α | UC | |
| Debiaerse | Vaccine against TNF-α consisting of a TNF-α derivative TNF-α kinoid | CD | |
| Effector T cells, B cells | Antigen specific Type 1 regulatory cells (OvaSave) | Autologous ova expanded regulatory T cells injected | CD |
| α4 integrin | AJM-300 | Blockade of α4 integrin | CD |
| α4 integrin | Natalizumab | Blockade of α4 integrin | Both |
| α4β7 integrin | Vedolizumab | Blockade of α4β7 integrin | Both |
| β7 integrin | Etrolizumab (aka rHuMab β7) | Anti-β7 integrin | UC |
Introduction
Inflammatory bowel disease (IBD), specifically Crohn disease (CD) and ulcerative colitis (UC), are autoimmune diseases whose incidence and prevalence are increasing worldwide. Over the last few decades, substantial progress has been made in understanding the pathophysiology of IBD, which has been translated into newer, more effective therapies (biologics) that have reduced flares, brought more patients into remission, and improved the quality of life of patients with IBD. IBD is considered to be an immune-mediated disease that involves a complex pattern/interplay of host genetics and environmental influences. Our knowledge of the immune system and its homeostatic imbalance is derived from mouse models of colitis and human studies involving clinical and laboratory experiments.
The immune system evolved in multicellular organisms/metazoans as a defense mechanism against pathogens like bacteria, protozoa, parasites, and fungi. The human immune system can be broadly categorized into innate and adaptive based on the differences in timing of the response and specificity. The immune system comes in contact with a foreign challenge, which could be food, commensal flora, microbial pathogens, and xenobiotics at different sites like the skin, mucous membrane of lungs, gastrointestinal tract, and so forth. The human gastrointestinal tract, with a total surface area roughly equal to that of a tennis court (400 m 2 ), serves as the largest area of interface with the external environment. The gut mucosal immune system, which interacts with this large antigenic load, thus, has the most varied immune cells in the body. In a disease-free host, there is a fine balance between a protective and deleterious response of the immune system, which becomes perturbed in patients with IBD. To understand these perturbations in IBD that produce a disease state, it is necessary to first understand how the intestinal immune system works. In this review, the authors divide their article into subsections of innate and adaptive immunity and link it with the currently identified abnormalities in these pathways in IBD. In addition, the authors have summarized in Table 1 the current and emerging therapies for IBD that target specific molecules in the immune system.
| Biologic Target | Antibody/Drug | Mechanism of Action | CD, UC, or Both |
|---|---|---|---|
| CCR9 | CCX282-B | Inhibition of CCR9 | CD |
| CCX 025 | Inhibition of CCR9 | CD | |
| IL-21 | PF 05230900 | IL-21 receptor antagonist | CD |
| IL-13 | QAX576 | IL-13 antagonist | CD |
| Anrukinzumab | IL-13 antagonist | UC | |
| Tralokinumab | IL-13 antagonist | UC | |
| IL-17 | Vidofludimus | Inhibitor of IL-17 A and IL-17F | Both |
| IL-12/23 | Ustekinumab | Blockade of IL-12/23 | CD |
| IL-18 | GSK1070806 | Blockade of soluble IL-18 | CD |
| IL-6 and IL-6R | Tocilizumab | Inhibitor of IL -6 | CD |
| PF04236921 | Inhibitor of IL -6 | CD | |
| IP-10 | MDX 1100 | Blockade of interferon-γ inducible protein (IP-10 or CXCL10) | UC |
| IRAK4/TRAF6/MyD88 | RDP58 | Disrupts IRAK4/TRAF6/MyD88 signaling and reduces production of proinflammatory cytokines | Both |
| JAK3 | Tofacitinib | Inhibition of JAK3 | Both |
| MAdCAM-1 | PF-547659 | Blocks MAdCAM-1 | Both |
| NF-κB | HE3286 | Synthetic steroid that modulates NF-κB activity | UC |
| NKG2D | NN8555 | Anti-NKG2D receptor monoclonal antibody | CD |
| PKC | AEB071/Sotrastaurin | PKC inhibitor | UC |
| T Cell | Laquinimod | Reduces IL-17 level and interferes with migration of T cells | CD |
| TLR | DIMS0150 | Blockade of Toll-like receptor | UC |
| BL-7040 | Blockade of Toll-like receptor | UC | |
| TNF-α | Infliximab | Neutralization of TNF-α | Both |
| Adalimumab | Neutralization of TNF-α | Both | |
| Certolizumab pegol | Neutralization of TNF-α | CD | |
| Golimumab | Neutralization of TNF-α | UC | |
| Debiaerse | Vaccine against TNF-α consisting of a TNF-α derivative TNF-α kinoid | CD | |
| Effector T cells, B cells | Antigen specific Type 1 regulatory cells (OvaSave) | Autologous ova expanded regulatory T cells injected | CD |
| α4 integrin | AJM-300 | Blockade of α4 integrin | CD |
| α4 integrin | Natalizumab | Blockade of α4 integrin | Both |
| α4β7 integrin | Vedolizumab | Blockade of α4β7 integrin | Both |
| β7 integrin | Etrolizumab (aka rHuMab β7) | Anti-β7 integrin | UC |
Innate intestinal immunity
Epithelial Barrier
The gastrointestinal tract has a continuous layer of single epithelial cells that are derived from a common progenitor LGR5+ intestinal stem cell. The epithelial cells comprise enterocytes (intestinal absorptive cells), goblet cells, neuroendocrine cells, Paneth cells, and microfold (M) cells. The epithelial cells are sealed with intercellular tight junctions that serve a barrier function and regulate the trafficking of macromolecules between the luminal environment and the host. The tight junctions are composed of a meshwork of proteins like occludin, claudin family members, and the junctional adhesion molecule, with zonulin as one of the physiologic modulators (of tight junctions) that controls intestinal permeability. The luminal surface is covered by a thick layer of mucus, which is produced by goblet cells. Mucus is rich in secreted immunoglobulin A (IgA) antibodies, proteins with antibacterial activity (ie, α- and β-defensins), and proteolytic and glycolytic enzymes that form the first line of defense against invasion by foreign pathogens. In spite of this barrier, gut bacteria and luminal antigens do enter the subepithelial lamina propria (an extracellular matrix compartment that contains a variety of immune and nonimmune cells), with some entering through unwanted breaks but most through the specialized, follicle-associated epithelium (FAE) that overlies the organized lymphoid tissue of the gut (gut-associated lymphoid tissue [GALT]). GALT consists of organized lymphoid compartments (Peyer patches, mesenteric lymph nodes, isolated lymphoid follicles, cryptopatches) and dispersed lymphoid cells in lamina propria and intraepithelial spaces. The FAE overlies the large lymphoid aggregates called Peyer patches and, compared with other parts of intestinal epithelium, is devoid of goblet cells and has a lower concentration of brush border enzymes. The FAE also contains unique highly specialized epithelial cells called M cells that actively pinocytose or endocytose selected luminal macromolecules into underlying lymphoid follicles, thus, facilitating transepithelial transport. M cells, in comparison with enterocytes, have irregular microvilli, decreased alkaline phosphatase activity, and no IgA secretory component. Immediately below the FAE is the subepithelial dome region that overlies the lymphoid follicle containing large numbers of dendritic cells (DCs). The antigens and microorganisms transported through M cells are captured by these DCs, which then present antigenic peptides to interfollicular T cells or naïve T cells within the mesenteric lymph nodes ( Fig. 1 ). Paneth cells are epithelial cells found at the base of the small intestinal crypt that can sense luminal microbiota and antigens to secrete antimicrobial peptides and contribute to innate host immunity.
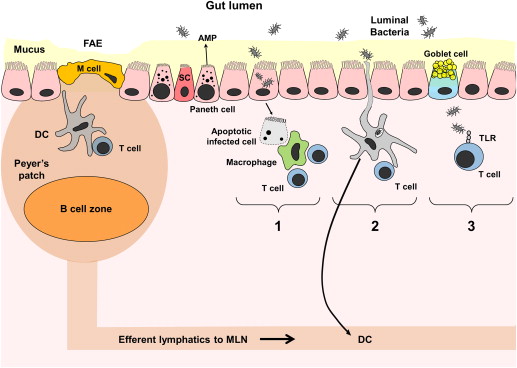
Innate Immune Cells
The cells of the innate immune system, in contrast to the adaptive immune system, have a rapid and less specific response to invading microorganisms or toxic macromolecules. This response is mediated by pathogen recognition receptors (PRR), which are membrane-bound (ie, Toll-like receptors [TLRs], C-type lectin receptor) or cytoplasmic (ie, nucleotide-binding oligomerization domain family members [NODs], retinoic acid–inducible gene 1–like receptor). These receptors are germ-line encoded, invariable, and predetermined to recognize a repertoire of bacterially/virally associated carbohydrate and lipid structures leading to the rapidity of response. The microbial molecules recognized by these receptors are called pathogen-associated molecular patterns (PAMPs) (eg, lipopolysaccharide and peptidoglycan). The PAMPs are highly conserved molecules on microbes as they are central to survival. These receptors also recognize endogenous or self-danger signals (eg, heat-shock proteins, uric acid), which are known as damage-associated molecular pattern molecules. The professional phagocytic cells (macrophages, dendritic cells and neutrophils), eosinophils, and lymphoid cells have immediate bactericidal activity; in the following paragraphs, the authors present data on these cells and their functions.
Macrophages
Macrophages are phagocytic cells present throughout the gastrointestinal mucosa, with the GALT containing the largest reservoir of macrophages within the adult mouse. Macrophages are derived from peripheral blood monocytes and are recruited to the intestine by chemoattractants, chemokines, and bacterial degradation products. Macrophages mature after migrating to intestinal mucosa, in contrast to neutrophils that are produced and lost in large numbers each day. In the noninflamed mucosa, macrophages differ from peripheral monocytes in their downregulated proinflammatory function, characterized by hyporesponsiveness to TLR ligands, diminished ability to prime adaptive immune responses, but still preserved capacity for phagocytosis and intracellular killing. However, in the setting of pathogen invasion and inflammation, intestinal macrophages freshly recruited from blood monocytes rapidly convert to a proinflammatory phenotype by ligation of their PRR. This proinflammatory phenotype is marked by abundant cytokine production (interleukin [IL]-1, IL-6, IL-8 and tumor necrosis factor [TNF]-alpha) and accessory cell function, instead of a primarily scavenger function. Once activated, the macrophages express receptors specific for opsonized particles and pathogens, complement, and common bacterial proteins (ie, mannose receptor, TLR, NOD). Pathogens are recognized by these receptors, which lead to their phagocytosis and subsequent intracellular killing. In addition, the activated macrophages secrete cytokines, which are proteins that affect the function of other cells in the absence of cell contact. Transforming growth factor beta (TGF-β), a cytokine produced by activated macrophages, is a potent chemoattractant for other macrophages and neutrophils and, hence, augments the recruitment of these cells to sites of inflammation. Cytokine secretion not only augments the phagocytic intracellular killing but also serves as a critical link between the 2 arms (ie, innate and adaptive of the immune system) and plays an important role in gut homeostasis and inflammation. For simplicity, macrophages had been broadly divided into inflammatory M1 and wound-healing M2; but recently, this view is being replaced by one of considerable plasticity of macrophages wherein they acquire distinct functions of host defense, wound healing, and immune regulation based on environmental cues they encounter. So they are considered a highly heterogeneous population of cells with a continuum of activation states.
DCs
DCs are phagocytic cells, which, like the macrophages, originate from blood monocytes or a common DC progenitor in the bone marrow. DCs have the most potent ability to initiate adaptive immune responses against pathogens. Current evidence supports that intestinal DCs continuously migrate in an immature or tolerogenic state, form scavenging apoptotic cells, and acquire antigens sampled either directly from the lumen or shuttled from the lumen through M cells (see Fig. 1 ). After the acquisition of antigen, DCs process it within, load the antigenic peptide onto major histocompatibility complex (MHC) class II molecules, and display this complex on its cell surface. DCs then migrate to the draining mesenteric lymph nodes where they present a particular MHC-peptide complex to T cells bearing the T cell receptor (TCR) specific for the antigen being presented. In a normal physiologic state, these antigen-loaded DCs (intestinal DCs) express low levels of costimulatory molecules and cytokines and, on contact with T cells, preferentially stimulate their differentiation into T regulatory cells (Tregs) that produce antiinflammatory cytokines IL-4, IL-10, and TGF-β. In contrast, when intestinal DCs are activated in a proinflammatory microenvironment, they migrate to T cell areas of the GALT, where they induce effector rather than tolerogenic T-cell responses. The DCs isolated from these sites express high levels of costimulatory molecules and adhesion molecules and produce large amounts of cytokines. The intestinal DCs can then induce the mucosal homing receptor α4β7 and the chemokine receptor CCR9 on T cells, suggesting that they can instruct a T cell to home back to the original site of the DC’s activation.
Atypical lymphocytes
The atypical lymphocytes γδT cells and natural killer T cells (NKT) are also a part of the innate immune response. T cells normally express alpha and beta TCR chains; but when they express gamma and delta TCR chains (γδT cells), they are considered atypical lymphocytes. These γδT cells only exist in significant numbers in the epithelial tissues and constitute greater than 10% of small intestinal intraepithelial lymphocytes, with the remaining majority being CD8+ T cells. The γδT cells, unlike the conventional T cells, do not depend on the thymus for development and do not recognize antigen in association with MHC class I or II. The γδT cells perform different functions, including acting as effectors against pathogens and tumors and as professional antigen-presenting cells (APCs); but their role depends on tissue distribution and the local microenvironment. NKT cells are a subset of T cells that mature in the thymus and recognize lipid antigen (presumably bacterial) presented on the MHC-like complex Cd1d. On activation, they secrete large quantities of proinflammatory cytokines and readily kill infected cells or tumor cells. NKT cells that produce large amounts of proinflammatory cytokine IL-13 have been found in the intestine of patients with UC and are also the mediators of inflammation in the oxazolone-induced murine model of colitis, which resembles UC.
Evidence for innate immune cellular dysfunction in the pathogenesis of IBD
There are multiple lines of evidence that indicate a major role for innate immune cells in the perpetuation of human chronic intestinal inflammation. The most recent meta-analysis of genome-wide association studies (GWAS) in IBD has implicated multiple susceptibility genes involved in innate mucosal defense (NOD2, CARD9, REL, SLC1A) and antigen presentation (ERAP2, LNPEP). Indeed, one of the strongest genetic associations to date with small bowel CD is the loss-of-function polymorphisms in the bacterial sensing gene CARD15/NOD2. The dysfunction secondary to NOD2 mutation has been reported in Paneth cells and monocytes. It seems that dysregulation in the detection of and/or responsiveness to enteric bacteria by innate immune and epithelial cells promotes chronic inflammation in this subgroup of patients. More evidence for their role rests in the efficacy of immune therapy targeted against proinflammatory cytokines produced by innate immune cells. Activated macrophages produce large quantities of TNF-α and IL-12, which are 2 cytokines responsible for the recruitment and activation of pathogenic effector T cells. The efficacy of anti–TNF-α therapy in IBD is well established, and promising anti–IL-12/23 studies are ongoing.
Another feature in patients with CD is a defective inflammatory response to injury and bacterial products, as evidenced by decreased neutrophil infiltration, lower production of proinflammatory interleukins (IL-8 and IL-1β), and reduced vascular flow as compared with healthy individuals. In addition to defective acute response, there is failure of clearance of bacteria and inflammatory debris, which indicates a disruption in the autophagy pathway. Autophagy (or self-eating) is a process that involves degradation and recycling of intracellular contents and removal of intracellular microbes and is mediated by lysosomes. The GWAS first pointed to the role of autophagy in CD pathogenesis, with many CD-associated genetic loci lying within the category of autophagy homeostatic function (eg, ATG16L1 and IRGM). Homozygosity for the ATG16L1 risk allele contributes to Paneth cell dysfunction in mice and humans, but mice do not develop spontaneous intestinal inflammation. Another pathway interconnected with autophagy and that affects intestinal epithelial cells of patients with IBD is unfolded protein response (UPR) from endoplasmic reticulum (ER) stress. Most of the secreted and membrane proteins enter the ER where they fold and assemble with only properly folded proteins exiting the ER via vesicles. When the unfolded proteins accumulate in the ER, it may lead to ER stress and activation of signaling pathways collectively known as UPR that help mitigate the stress. Not only the intestines of patients with IBD have evidence of endoplasmic reticulum (ER) stress but multiple ER stress-related genes like XBP1 have been associated with IBD. Genetic deletion of XBP1 in the intestinal epithelial cells of mice results in the depletion of Paneth cells, inflammatory hyperresponsiveness to TNF-alpha, and spontaneous enteritis.
Evidence for barrier dysfunction and epithelial injury in the pathogenesis of IBD
Numerous studies have shown that disruption of the epithelial barrier may either initiate or perpetuate chronic intestinal inflammation. Abnormal intestinal permeability has been established among patients with CD and their healthy first-degree relatives and may represent a primary abnormality predisposing to excessive antigen uptake, continuous immune stimulation, and eventually mucosal inflammation. Animal models like SAMP1/YitFc have increased intestinal permeability before the development of spontaneous ileitis, and GWAS studies have identified as a CD risk locus MUC19, whose product comprises the intestinal mucous layer. Barrier dysfunction has been directly observed by confocal endomicroscopy in a cohort of patients with IBD, and this dysfunction is predictive of IBD relapse within 12 months of microscopy. The etiologic factors for barrier dysfunction in CD may be environmental or genetic. Diet and nonsteroidal antiinflammatory drugs can affect gut permeability and are variably associated with IBD. Furthermore, polymorphic variants in several IBD-associated genes (DLG5, JAK2, HNF4A) seem to primarily affect epithelial permeability and may lead to inappropriate exposure of the mucosal immune system to luminal antigens. Additionally, epithelial cell death, particularly of Paneth cells, has been demonstrated to induce terminal ileal inflammation in mice with conditional deletion of caspase-8 in the intestinal epithelium and is associated with CD in humans.
Related posts:
 Update on Anti-Tumor Necrosis Factor Agents in Crohn Disease
Update on Anti-Tumor Necrosis Factor Agents in Crohn Disease
 An Update on Anti-TNF Agents in Ulcerative Colitis
An Update on Anti-TNF Agents in Ulcerative Colitis
 Lymphocyte Homing Antagonists in the Treatment of Inflammatory Bowel Diseases
Lymphocyte Homing Antagonists in the Treatment of Inflammatory Bowel Diseases
 Biologics of IBD
The Use of Biologic Agents in Pregnancy and Breastfeeding
Update on Janus Kinase Antagonists in Inflammatory Bowel Disease
Biologics of IBD
The Use of Biologic Agents in Pregnancy and Breastfeeding
Update on Janus Kinase Antagonists in Inflammatory Bowel Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


