Lymphocyte homing antagonists represent promising therapeutic agents for the treatment of idiopathic inflammatory bowel disease (IBD). Several critical molecules involved in the recruitment of inflammatory cells in the intestine, including integrins and chemokine receptors, have been successfully targeted for the treatment of IBD. These agents have shown great promise for the induction and maintenance of remission for both Crohn disease and ulcerative colitis. This article discusses currently approved prototypic agents for the treatment of IBD (natalizumab, anti-α4 integrin; vedolizumab, anti-α4β7 integrin), and several other agents in the same class currently under development.
Key points
- •
Inhibiting the interactions between adhesion molecules expressed on inflamed intestinal endothelium and lymphocyte integrins has proved to be a successful therapeutic strategy for the management of chronic inflammatory bowel disease (IBD).
- •
Targeting specifically the MAdCAM-1/α4β7 interactions with vedolizumab and, possibly, AMG 181 is likely to be effective and safe treatment approach for refractory IBD patients.
- •
Future studies need to address the use of these agents in the treatment of fistulizing Crohn disease phenotypes and extraintestinal manifestation of IBD.
- •
Subgroup analysis may identify genetic, serologic, and clinical parameters that may predict response to adhesion molecule inhibition and the best timing for therapeutic intervention.
- •
Combination therapeutic strategies with immunomodulators or even anti–tumor necrosis factor agents in a selective refractory group of IBD patients should also be entertained.
Introduction
Inflammatory bowel diseases (IBD) comprise a heterogeneous group of intestinal inflammatory disorders characterized by periods of disease exacerbation and remission, and are broadly classified into ulcerative colitis (UC) and Crohn disease (CD). Their pathogenesis is still unknown, but they are thought to develop as a result of a dysregulated immune response to certain gut bacterial antigens in the context of genetic susceptibility, certain environmental triggers, and gut dysbiosis. Histopathologically, IBD is characterized by the extent and distribution of mucosal architectural abnormality, the cellularity of the lamina propria, and the cell types present, but with significant overlap between the 2 diseases. Disease activity can be reflected by neutrophil granulocyte infiltration and epithelial cell damage. Characteristics of disease that could interfere with response to medical therapy, whether to traditional medications or to biological agents (including adhesion molecule inhibitors), include early disease or long-standing disease, which could reflect differences in inflammatory cell infiltrate and their immunologic characteristics.
Traditional therapies of IBD, such as 5-aminosalicylic acid, corticosteroids, and immunomodulators, have been shown to modulate the intestinal immune responses to some degree in a nonspecific manner. The introduction of biological agents for the treatment of IBD has targeted specific pathways of inflammation, including proinflammatory cytokines such as tumor necrosis factor (TNF)-α, with remarkable long-term beneficial effects that may alter the natural history of IBD. Such treatment approaches include the introduction of monoclonal antibodies targeting TNF-α, such as infliximab, adalimumab, certolizumab pegol, and golimumab. However, despite the remarkable effectiveness of these agents, they may lose efficacy over time or rarely produce treatment-related side effects. Therefore, alternative therapeutic agents with a different mode of action are urgently needed.
Active IBD is characterized by the recruitment of large numbers of granulocytes, lymphocytes, and macrophages into the gastrointestinal mucosa. The recruitment of leukocytes to peripheral tissues is a highly coordinated, multistep process ( Fig. 1 ). As they circulate at high speed through the postcapillary vessels, a highly coordinated sequential adhesion pathway is activated, which consists of the capture/tethering, rolling, activation, adhesion, and migration through the vascular wall, and eventual transmigration to the tissue. These infiltrating leukocytes may perpetuate the inflammatory process through the secretion of proinflammatory cytokines, further endothelial cell activation, and upregulation of adhesion molecules and enhancement of inflammatory cell recruitment. Further tissue damage may result from the release of proteases by the infiltrating leukocytes. The process of intestinal leukocyte infiltration is regulated by the expression of integrins and chemokine receptors on leukocytes, and adhesion molecules such as intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and mucosal addressin cell adhesion molecule 1 (MAdCAM-1) expressed on endothelial cells (see Fig. 1 ).
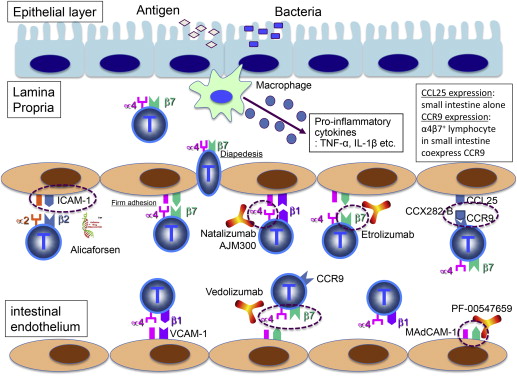
Integrins comprise a family of α,β heterodimeric transmembrane receptors that are constitutively expressed and mediate the attachment of cells to the extracellular matrix (ECM), but can also take part in specialized cell-cell interactions. The integrin family consists of at least 24 different forms representing the combination of 18 α subunits and 8 β subunits. These integrins are expressed on the surface of leukocytes and require activation to bind their own specific ligand. The α subunit determines integrin ligand specificity, and the β subunit connects to the cytoskeleton and affects multiple signaling pathways. Integrins involved in the T-cell migration include leukocyte function–associated antigen 1 (LFA-1 or α2β2) and the 2 α4 integrins (α4β1 and α4β7). LFA-1 is expressed on neutrophils, and interacts with ICAM-1, which is expressed on leukocytes, dendritic cells, fibroblasts, epithelial cells, and endothelial cells. The α4β1 integrin is expressed on most leukocytes but not on neutrophils, and interacts with VCAM-1. The α4β7 integrin is expressed on the lymphocytes in the gut-associated lymphoid tissue, and interacts with MAdCAM-1. This ligand is expressed on endothelial venules in the small intestine and the colon, especially in the Peyer patches.
Proinflammatory cytokines such as TNF-α and interleukin (IL)-1β upregulate the expression of gut-expressed adhesion molecules such as MAdCAM-1, ICAM-1, and VCAM-1. These adhesion molecules are overexpressed in IBD, especially in the active phase of the disease. Cultured supernatants from colonic biopsies taken from UC and CD patients induce upregulation of ICAM-1 and E-selectin, and there is increased expression of various endothelial adhesion molecules by immunohistochemistry from colonic biopsies in patients with IBD. Moreover, it has been shown that the proportion of venular endothelium within the lamina propria that expresses MAdCAM-1 at inflammatory foci associated with UC and CD is increased compared with normal tissues, and MAdCAM-1 is not detected in most normal or inflamed extraintestinal tissues, including those at other mucosal surfaces. Similarly, VCAM-1 is also overexpressed in the colonic IBD mucosa in comparison with normal controls by immunohistochemistry.
Introduction
Inflammatory bowel diseases (IBD) comprise a heterogeneous group of intestinal inflammatory disorders characterized by periods of disease exacerbation and remission, and are broadly classified into ulcerative colitis (UC) and Crohn disease (CD). Their pathogenesis is still unknown, but they are thought to develop as a result of a dysregulated immune response to certain gut bacterial antigens in the context of genetic susceptibility, certain environmental triggers, and gut dysbiosis. Histopathologically, IBD is characterized by the extent and distribution of mucosal architectural abnormality, the cellularity of the lamina propria, and the cell types present, but with significant overlap between the 2 diseases. Disease activity can be reflected by neutrophil granulocyte infiltration and epithelial cell damage. Characteristics of disease that could interfere with response to medical therapy, whether to traditional medications or to biological agents (including adhesion molecule inhibitors), include early disease or long-standing disease, which could reflect differences in inflammatory cell infiltrate and their immunologic characteristics.
Traditional therapies of IBD, such as 5-aminosalicylic acid, corticosteroids, and immunomodulators, have been shown to modulate the intestinal immune responses to some degree in a nonspecific manner. The introduction of biological agents for the treatment of IBD has targeted specific pathways of inflammation, including proinflammatory cytokines such as tumor necrosis factor (TNF)-α, with remarkable long-term beneficial effects that may alter the natural history of IBD. Such treatment approaches include the introduction of monoclonal antibodies targeting TNF-α, such as infliximab, adalimumab, certolizumab pegol, and golimumab. However, despite the remarkable effectiveness of these agents, they may lose efficacy over time or rarely produce treatment-related side effects. Therefore, alternative therapeutic agents with a different mode of action are urgently needed.
Active IBD is characterized by the recruitment of large numbers of granulocytes, lymphocytes, and macrophages into the gastrointestinal mucosa. The recruitment of leukocytes to peripheral tissues is a highly coordinated, multistep process ( Fig. 1 ). As they circulate at high speed through the postcapillary vessels, a highly coordinated sequential adhesion pathway is activated, which consists of the capture/tethering, rolling, activation, adhesion, and migration through the vascular wall, and eventual transmigration to the tissue. These infiltrating leukocytes may perpetuate the inflammatory process through the secretion of proinflammatory cytokines, further endothelial cell activation, and upregulation of adhesion molecules and enhancement of inflammatory cell recruitment. Further tissue damage may result from the release of proteases by the infiltrating leukocytes. The process of intestinal leukocyte infiltration is regulated by the expression of integrins and chemokine receptors on leukocytes, and adhesion molecules such as intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and mucosal addressin cell adhesion molecule 1 (MAdCAM-1) expressed on endothelial cells (see Fig. 1 ).
Integrins comprise a family of α,β heterodimeric transmembrane receptors that are constitutively expressed and mediate the attachment of cells to the extracellular matrix (ECM), but can also take part in specialized cell-cell interactions. The integrin family consists of at least 24 different forms representing the combination of 18 α subunits and 8 β subunits. These integrins are expressed on the surface of leukocytes and require activation to bind their own specific ligand. The α subunit determines integrin ligand specificity, and the β subunit connects to the cytoskeleton and affects multiple signaling pathways. Integrins involved in the T-cell migration include leukocyte function–associated antigen 1 (LFA-1 or α2β2) and the 2 α4 integrins (α4β1 and α4β7). LFA-1 is expressed on neutrophils, and interacts with ICAM-1, which is expressed on leukocytes, dendritic cells, fibroblasts, epithelial cells, and endothelial cells. The α4β1 integrin is expressed on most leukocytes but not on neutrophils, and interacts with VCAM-1. The α4β7 integrin is expressed on the lymphocytes in the gut-associated lymphoid tissue, and interacts with MAdCAM-1. This ligand is expressed on endothelial venules in the small intestine and the colon, especially in the Peyer patches.
Proinflammatory cytokines such as TNF-α and interleukin (IL)-1β upregulate the expression of gut-expressed adhesion molecules such as MAdCAM-1, ICAM-1, and VCAM-1. These adhesion molecules are overexpressed in IBD, especially in the active phase of the disease. Cultured supernatants from colonic biopsies taken from UC and CD patients induce upregulation of ICAM-1 and E-selectin, and there is increased expression of various endothelial adhesion molecules by immunohistochemistry from colonic biopsies in patients with IBD. Moreover, it has been shown that the proportion of venular endothelium within the lamina propria that expresses MAdCAM-1 at inflammatory foci associated with UC and CD is increased compared with normal tissues, and MAdCAM-1 is not detected in most normal or inflamed extraintestinal tissues, including those at other mucosal surfaces. Similarly, VCAM-1 is also overexpressed in the colonic IBD mucosa in comparison with normal controls by immunohistochemistry.
Preventing leukocyte infiltration by blocking adhesion receptors
Inhibition of leukocyte trafficking to the gut mucosa during the inflammatory process in IBD has become the second major therapeutic target of biological agents after the introduction of anticytokine agents in the development of IBD therapeutics. Several critical steps outlined in Fig. 1 that are involved in the recruitment of inflammatory cells to the intestinal mucosa can be targeted for therapeutic purposes. The α4β7 integrin is highly expressed on a subpopulation of CD4 + CD45RA – memory T cells, which have been shown to preferentially home to the gut. The ligand mediating α4β7 T-cell gut-homing is MAdCAM-1, which is selectively expressed in the gut endothelium and is upregulated in the chronically inflamed small and large intestines of patients with IBD.
The predominant targets of the novel biological agents in this group are the integrins α4β1, α4β7, and α2β2, which interact with VCAM-1, MAdCAM-1, and ICAM-1, respectively, to mediate the interactions between leukocytes and endothelial cells. The agents that target the integrins include the monoclonal antibodies natalizumab (anti-α4 integrin), vedolizumab (MLN-02, anti-α4β7 integrin), AMG181 (anti-α4β7 integrin), etrolizumab (RG7413, rhuMAb β7, anti-β7 integrin), which targets both α4β7 and αEβ7, and PF-00547659 (anti-MAdCAM-1). AJM-300 (Ajinomoto Pharmaceuticals, Tokyo, Japan) is a small-molecule inhibitor of the α4 integrin subunit. Other molecules that aim to prevent leukocyte infiltration include alicaforsen (ISIS-2302), an antisense oligonucleotide against ICAM-1 messenger RNA. Vercirnon (CCX282-B, GSK-1605786) is a small molecule that targets the chemokine receptor CCR9, the key chemokine receptor in the targeting of leukocytes to the intestinal mucosa (see Fig. 1 , Table 1 ).
| Drug | Description | Developer | Target | Indication | Clinical Status |
|---|---|---|---|---|---|
| Tysabri | Humanized IgG4 mAb | Biogen Idec (Cambridge, MA) | α4β1 integrin, α4β7 integrin | Multiple sclerosis, Crohn disease | Approved (FDA) |
| Entyvio | Humanized IgG1 mAb | Takeda Pharmaceuticals (Deerfield, IL) | α4β7 integrin | Crohn disease | Registration (in USA) |
| AMG-181 | Fully human IgG2 mAb | AstraZeneca (London, UK)/Amgen (Thousand Oaks, CA) | α4β7 integrin | Crohn disease, ulcerative colitis | Phase 2 |
| Etrolizumab (rhuMAb β7, RG7413) | Humanized IgG1 mAb | Genentech (South San Francisco, CA) | α4β7 integrin, αEβ7 integrin | Ulcerative colitis | Phase 2 |
| PF-00547659 | Fully human IgG2k mAb | Pfizer (New York, NY, USA) | MAdCAM-1 | Crohn disease, ulcerative colitis | Phase 2 |
| AJM300 | Oral small-molecule prodrug | Ajinomoto Pharmaceuticals (Tokyo, Japan) | α4 integrin | Ulcerative colitis, Crohn disease | Phase 2 |
| Vercirnon (CCX282-B) | Oral small molecule | ChemoCentryx (Mountain View, CA)/GSK (Brentwood, Middlesex, UK) | CCR9 | Crohn disease | Phase 3 (on hold) |
Current antiadhesion therapies for IBD
Antiadhesion Therapies for IBD
Natalizumab
Natalizumab (Tysabri; Biogen-Idec, Cambridge, MA, USA) is a humanized immunoglobulin G4 (IgG4) monoclonal antibody against α4 integrins. Previous randomized studies have suggested that natalizumab may be effective as induction therapy for patients with moderately to severely active CD. The first study of natalizumab was reported by Gordon and colleagues, and focused on the evaluation of the safety and efficacy of natalizumab in patients with mildly to moderately active CD.
Ghosh and colleagues conducted a double-blind, placebo-controlled trial of natalizumab in 248 patients with moderate to severe CD. Patients were randomly assigned to receive 1 of 4 treatments: 2 infusions of placebo; 1 infusion of 3 mg natalizumab per kg body weight, followed by placebo; 2 infusions of 3 mg natalizumab per kg; or 2 infusions of 6 mg natalizumab per kg. The highest remission rate was 44% and the highest response rate was 71% at week 6 in the group given 2 infusions of 3 mg/kg.
Patients with moderate to severe disease (Crohn Disease Activity Index [CDAI] scores ≥220 to ≤450) were enrolled in the phase 3 induction trial, Efficacy of Natalizumab as Active Crohn Therapy (ENACT-1) (N = 905), and received 3 infusions of natalizumab (300 mg) or placebo over 8 weeks. Response was defined as a reduction of 70 or more points from week 0 in the CDAI score, whereas remission was defined as a CDAI score of less than 150 points. A clinical response at week 10 was observed in 56% of patients treated with natalizumab and 49% of patients with placebo ( P = .051), and remission obtained at week 10 in 37% of natalizumab-treated patients and 30% of patients with placebo ( P = .124). Although there were no significant differences among the 2 treatment groups, analysis in subgroups of patients, including those with active inflammation and a baseline C-reactive protein (CRP) concentration above the upper limit of normal range (>2.87 mg/L), demonstrated clinically and statistically significant differences in response and remission rates. Among the patients in the ENACT-1 trial with CRP concentrations above the upper limit of normal (n = 660; 73% of the total registered patients), 58% of natalizumab-treated patients and 45% of patients with placebo had a clinical response at week 10 ( P = .007) while 40% and 28%, respectively, had clinical remission ( P = .014).
Targan and colleagues performed a randomized, placebo-controlled trial that evaluated the efficacy of natalizumab induction therapy in patients with moderately to severely active CD and active inflammation characterized by elevated CRP concentrations (Efficacy of Natalizumab in Crohn’s Disease Response and Remission [ENCORE] trial). In this trial, 509 patients with moderately to severely active CD were enrolled; 48% of natalizumab-treated patients had a sustained response at week 8 through week 12 compared with 32% of patients treated with placebo ( P <.001), while 26% and 16%, respectively, had sustained remission ( Fig. 2 ) ( P = .002).
The Evaluation of Natalizumab as Continuous Therapy (ENACT-2) trial evaluated the efficacy of natalizumab to maintain clinical response. In ENACT-2, 339 patients who had a response to natalizumab in the ENACT-1 trial were randomly reassigned to receive 300 mg of natalizumab or placebo every 4 weeks through week 56. The primary outcome was a sustained response through week 36. A secondary outcome was disease remission (CDAI score<150). Continuing natalizumab in the second trial resulted in higher rates of sustained response (61% vs 28%, P <.001) and remission (44% vs 26%, P = .003) through week 36 than did switching to placebo.
A pilot study evaluating the efficacy of natalizumab in UC patients was reported in 2002. A significant decrease in the median Powell-Tuck score was observed at 2 and 4 weeks postinfusion (7.5 and 6, respectively) compared with the median baseline score of 10. Five of 10 patients achieved a good clinical response at 2 weeks and 1 more patient by 4 weeks, defined by a Powell-Tuck score of 5 or less. Significant improvements in quality-of-life scores were found at week 4. The median CRP at 2 weeks (6 mg/L) was also lower than that pretreatment (16 mg/L); however, rescue medication was required by 2 (20%), 3 (30%), and 8 (80%) patients by weeks 2, 4, and 8, respectively (median, 34 days; range, 8–43 days).
Safety of natalizumab
The main serious adverse event that has been reported with the use of natalizumab in multiple sclerosis (MS) and CD studies and postmarketing experience is the development of progressive multifocal leukoencephalopathy (PML), a demyelinating disease of the white matter of the brain caused by lytic infection of oligodendrocytes with the human polyomavirus, John Cunningham virus (JCV). Although most PML cases occur in severely immunosuppressed individuals, with human immunodeficiency virus 1 infection as the predominant factor, PML has been increasingly diagnosed in patients treated with biological therapies such as anti–LFA-1 (efalizumab [Raptiva] for severe forms of plaque psoriasis) that prevents extravasation of inflammatory T cells into tissues, or anti-CD20 (rituximab [Rituxan] for hematologic malignancies and rheumatoid arthritis) that depletes peripheral circulating B cells. Mutations of JCV capsid viral protein 1 (VP1), the capsid protein involved in binding to sialic acid cell receptors, might favor PML onset. JCV is often acquired during childhood. Most adults have been infected with JCV but do not develop the disorder. The virus seems to remain inactive until immune suppression allows it to be reactivated and start to multiply. During clinical trials of natalizumab in more than 2000 MS and 1000 CD patients, PML was diagnosed in 2 MS patients and one CD patient in 2005. As of April 3, 2014, among approximately 123,000 patients who have received natalizumab worldwide in the postmarketing setting, 454 have developed PML, and the estimated incidence of PML is 3.6 per 1000 patient-years (Biogen Idec, personal communication, 2014). Three factors that are known to increase the risk of PML in natalizumab-treated patients have been identified: (1) longer treatment duration, especially beyond 2 years; (2) prior treatment with an immunosuppressive agent; and (3) the presence of anti-JCV antibodies. Patients who are anti-JCV antibody positive have a higher risk for developing PML ( Table 2 ).
| Anti-JCV Antibody Negative a | Tysabri Exposure (mo) b | Anti-JCV Antibody Positive c | |
|---|---|---|---|
| No Prior Immunosuppressant Use | Prior Immunosuppressant Use | ||
| <1/1000 | 1–24 | <1/1000 | 1/1000 |
| 25–48 | 3/1000 | 13/1000 | |
| 49–72 | 7/1000 | 9/1000 | |
Related posts:
 Anti–Tumor Necrosis Factor-α Monotherapy Versus Combination Therapy with an Immunomodulator in IBD
Anti–Tumor Necrosis Factor-α Monotherapy Versus Combination Therapy with an Immunomodulator in IBD
 Update on Anti-Tumor Necrosis Factor Agents in Crohn Disease
Update on Anti-Tumor Necrosis Factor Agents in Crohn Disease
 Pharmacokinetics of Biologics and the Role of Therapeutic Monitoring
Pharmacokinetics of Biologics and the Role of Therapeutic Monitoring
 Biologics of IBD
The Use of Biologic Agents in Pregnancy and Breastfeeding
Update on Janus Kinase Antagonists in Inflammatory Bowel Disease
Biologics of IBD
The Use of Biologic Agents in Pregnancy and Breastfeeding
Update on Janus Kinase Antagonists in Inflammatory Bowel Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


