ESSENTIALS OF DIAGNOSIS
ESSENTIALS OF DIAGNOSIS
Approximately 3–5% of colorectal cancers (CRCs) are caused by inherited gene mutations associated with hereditary nonpolyposis colorectal cancer (HNPCC) (Lynch syndrome) or adenomatous polyposis syndromes.
Genetic testing is clinically available for several hereditary gastrointestinal cancer syndromes and can be used to guide cancer screening recommendations.
The majority of cases of gastrointestinal cancer are believed to be sporadic events; however, inherited factors play a role in development of some tumors, with an estimated 5% being attributable to a single gene mutation. Hereditary gastrointestinal cancer syndromes convey a markedly increased risk for developing cancer and require specific strategies for diagnosis and management.
Identification of hereditary gastrointestinal cancer syndromes requires a thorough evaluation of patients’ personal and family history of cancer. Clinical genetic testing can be useful in confirming the diagnosis of certain hereditary cancer syndromes and guiding cancer screening for family members. Tables 23–1 and 23–2 summarize clinical characteristics and cancer screening recommendations for the hereditary gastrointestinal cancer syndromes discussed in detail in this chapter.
| Syndrome | Clinical Features | Gene(s) | Genetic Testing Available |
|---|---|---|---|
| Lynch syndrome (HNPCC) | High risk for colorectal and extracolonic cancers (endometrial, ovarian, urinary tract, sebaceous skin tumors) Young ages at cancer diagnosis Accelerated adenoma—carcinoma sequence CRC tumors demonstrate microsatellite instability | DNA mismatch repair (MMR) genes (MLH1, MSH2, MSH6, PMS2, TACSTD1/EPCAM) | Yes |
| Familial adenomatous polyposis (FAP) | 100–1000 colorectal adenomas appearing in second or third decade CRC risk 100% if colon is not removed Increased risk for duodenal and ampullary adenocarcinoma Risk of desmoid tumors | Tumor-suppressor gene APC or base excision repair gene MutYH biallelic mutations (autosomal recessive) | Yes |
| Attenuated polyposis/MutYH-associated polyposis (MAP) | 10–100 colorectal adenomas High risk for CRC ± Upper gastrointestinal adenomas | Tumor-suppressor gene APC Base excision repair gene MutYH (autosomal recessive) | Yesa |
| Peutz-Jeghers syndrome (PJS) | Hamartomatous polyps in GI tract (symptoms of bleeding, intussusception) Pigmented lesions on lips Increased risk for breast cancer, pancreatic cancer, sex cord tumors | Tumor-suppressor gene STK11 | Yesa |
| Juvenile polyposis | 5+ juvenile polyps in GI tract Family history of GI cancers Increased risk for CRC and other GI cancers Associated with congenital cardiac abnormalities | Tumor-suppressor genes SMAD4, BMPR1A | Yesa |
| Cowden syndrome | Hamartomatous polyps Increased risk for breast and thyroid cancers Macrocephaly | PTEN | Yesa |
| Hereditary diffuse gastric cancer | Diffuse infiltrative adenocarcinoma of the stomach (linitis plastica) Increased risk for breast cancer | Tumor suppressor gene e-cadherin/CDH1 | Yesa |
| Familial pancreatic cancer | Three or more family members with pancreatic cancer May be present as part of other hereditary cancer syndromes, eg, breast/ovarian cancer (HBOC), melanoma (FAMMM), or colorectal/gynecologic cancers (Lynch syndrome) | BRCA1/2 (HBOC) P16/CDKN2A (FAMMM) MMR genes (Lynch syndrome) PRSS1, SPINK1 (hereditary pancreatitis) | Yesa |
| Syndrome | Screening Test | Frequency |
|---|---|---|
| Lynch syndrome/HNPCC |
|
|
| Familial adenomatous polyposis (FAP) |
|
|
| Attenuated polyposis (AFAP) |
|
|
| MutYH-associated polyposis (MAP); |
|
|
| Peutz-Jeghers syndrome (PJS) |
|
|
| Juvenile polyposis |
|
|
| Cowden syndrome |
|
|
| Hereditary diffuse gastric cancer |
|
|
| Familial pancreatic cancer |
|
|
[PubMed: 20420945]
[PubMed: 20065170]
[PubMed: 25645574]
HEREDITARY COLORECTAL CANCER SYNDROMES
CRC is the most common gastrointestinal malignancy, with more than 140,000 cases diagnosed each year in the United States. Although most CRC patients do not have a striking family history of CRC, approximately 30% report having one or more family members with a diagnosis of CRC. The lifetime risk of developing CRC is approximately 5% for the average American; however, individuals who have a first-degree relative with CRC have a twofold higher risk for developing colorectal neoplasia compared with individuals who have no family history of CRC. For individuals with numerous relatives with CRC, the cancer risk may be markedly higher; those who have inherited mutations in genes involved in mismatch repair (MMR) or tumor suppression have a lifetime risk of CRC of 70–100% in the absence of medical intervention.
Identification of patients at risk for hereditary CRC syndromes relies on careful family history evaluation, because many individuals may not demonstrate a characteristic phenotype. Cancer risk stratification for every patient should involve eliciting a family history of cancers, including type of cancer and age of onset as well as family history of colorectal adenomas. Individuals whose family history includes multiple individuals with cancer, individuals diagnosed with two or more primary cancers, or with tumors diagnosed at young ages, should undergo more extensive family history evaluation of first-, second-, and third-degree relatives to determine whether there is evidence of an autosomal-dominant or autosomal-recessive pattern of inheritance.
Lynch syndrome, also known as HNPCC, is the most common hereditary CRC syndrome and is estimated to account for approximately 3% of CRC cases. This syndrome was first described by Dr Henry Lynch in families in which multiple cases of CRC were diagnosed at young ages. The original Lynch syndrome families were identified as having three or more cases of CRC with at least one diagnosed before age 50, as described by the Amsterdam criteria; however, additional studies have demonstrated that the cancer spectrum in these families includes other cancers, such as gastrointestinal, gynecologic, urinary tract, and sebaceous neoplasms of the skin. Because as many as 50% of families with Lynch syndrome do not meet the classic Amsterdam criteria, clinical diagnostic criteria have been expanded and modified to improve diagnostic sensitivity. As outlined by the Revised Bethesda guidelines (Table 23–3), Lynch syndrome should be suspected in families that have multiple relatives affected with CRC or related extracolonic tumors, or both, and in individuals who are diagnosed with CRC at a young age, have synchronous or metachronous colorectal cancers, or develop multiple Lynch-associated tumors. Figure 23–1 shows a pedigree of a family fulfilling criteria for Lynch syndrome.
|
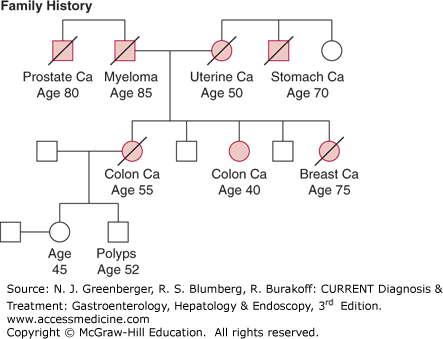
CRC is usually the predominant cancer in most families with Lynch syndrome. Initial studies suggested that the mean age of onset of CRC is 44 years; however, there is wide variation in ages of diagnosis among families. Although colonic tumors in Lynch syndrome are often right-sided, many patients develop tumors in the left colon and rectum. Synchronous or metachronous tumors are common, and any individual diagnosed with two primary colon cancers warrants evaluation for Lynch syndrome. Although Lynch syndrome is also referred to as HNPCC, most of the colorectal cancers do appear to arise from adenomatous polyps, although these polyps may be few in number and are often small and flat. The colorectal adenoma–carcinoma sequence appears to be accelerated in Lynch syndrome, and there are many reports of tumors arising within 3 years of a normal colonoscopy. Histopathologic features seen in colon tumors in Lynch syndrome include tumor infiltrating lymphocytes, Crohn-like inflammatory reaction, signet ring cells, and most exhibit features of defective DNA MMR (see Genetic Features).
Endometrial (uterine) cancer is the second most common cancer described in Lynch syndrome families; however in some families, cases of endometrial cancers may outnumber CRC. Women with Lynch syndrome have a 40–60% lifetime risk for developing this malignancy, which is an unusual cancer in the general population. The lifetime risks for developing other Lynch-associated cancers, such as urinary tract cancers, ovarian cancer, and other gastrointestinal cancers (stomach, pancreas, small intestine), are also increased for individuals with Lynch syndrome and are estimated to be between 10% and 20%. Brain tumors (eg, glioblastomas and astrocytomas) have been described in the Turcot syndrome variant of Lynch syndrome. Cutaneous sebaceous adenomas and sebaceous carcinomas are rare skin tumors seen in the Muir-Torre variant, and it is currently recommended that any individual affected with sebaceous neoplasms of the skin undergo evaluation for Lynch syndrome regardless of his or her family history.
The increased predisposition to developing cancer in Lynch syndrome is the result of autosomal dominantly inherited mutations in genes involved in DNA MMR. Mutations in the genes hMLH1 and hMSH2 account for more than 80% of the identified MMR alterations in Lynch syndrome families. Mutations in the MMR gene hMSH6 have been identified in approximately 10% of Lynch syndrome families, and in hPMS2 in rare families. Mutations in the gene TACSTD1/EPCAM, which regulates expression of MSH2, have also been found in families with a clinical diagnosis of Lynch syndrome.
The protein products of MMR genes are involved in identifying and repairing errors that arise during DNA replication. In the setting of defective MMR gene function, these errors accumulate in segments of DNA containing repeated sequences known as microsatellites. DNA errors that disrupt the function of genes involved in growth regulation can lead to the development of tumors. Approximately 85% of the colorectal tumors in Lynch syndrome patients demonstrate high levels of microsatellite instability (MSI), a characteristic of defective MMR gene function. Immunohistochemical (IHC) analysis of colorectal tumors for expression of MMR proteins MLH1, MSH2, MSH6, and PMS2 frequently reveals loss of staining of the protein corresponding to the gene with the mutation. Since only 15% of sporadic CRC tumors demonstrate high levels of MSI, testing CRC tumors for MSI and loss of expression of MMR proteins has been proposed as a cost-effective strategy to identify which patients with CRC may be at risk for Lynch syndrome and require additional genetic evaluation.
Approximately 60% of families that meet classic Amsterdam criteria are found to have germline mutations in one of the DNA MMR genes. However, classic Amsterdam families with multiple CRC diagnoses may include not only Lynch syndrome families, but also families with familial colorectal cancer syndrome X, which may or may not have a genetic basis. Like Lynch syndrome, familial colorectal cancer syndrome X families have multiple cases of CRC with apparent autosomal-dominant pattern of inheritance; however, the colorectal tumors in patients with familial colorectal cancer syndrome X do not have features of MSI and affected individuals do not appear to be at increased risk for extracolonic cancers.
The high lifetime risk of colorectal and other extracolonic cancers, the accelerated progression of adenomas to adenocarcinomas, and the young age of onset of colorectal neoplasia require specialized strategies for cancer prevention.
Individuals who are at risk for Lynch syndrome should begin having colonoscopies at age 20–25, with repeat examinations every 1–2 years, although earlier initiation of screening is recommended 2–5 years before the youngest age of diagnosis of CRC in the family if diagnosed before age 25. The need for a shorter interval between examinations became evident from European studies that demonstrated a reduction in CRC mortality for individuals who had colonoscopies every 3 years; however, cancers were still detected during that screening interval. The endoscopist should be vigilant for small or flat lesions, which may be associated with higher malignant potential in Lynch syndrome patients than in the general population.
There is growing, but inconclusive, evidence that the use of aspirin is beneficial at reducing the risk of colon cancer in Lynch syndrome patients, but not without significant risk of adverse events. Ongoing research is underway to establish the optimal dose and duration of aspirin treatment, and other novel agents are being studied in this population.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


