CHAPTER 57 Hereditary, Familial, and Genetic Disorders of the Pancreas and Pancreatic Disorders in Childhood
DEFINITIONS AND TERMINOLOGY
There are a growing number of pancreatic disorders with a known genetic basis. However, there is no consensus terminology to distinguish disorders of different etiology that have identical end-stage pathology. Acute pancreatitis represents an event triggered by sudden pancreatic injury that is followed by sequential inflammatory responses (see Chapter 58). Chronic pancreatitis, on the other hand, is a process that usually begins with recurrent acute pancreatitis and ends with immune-mediated destruction of the pancreas and widespread glandular fibrosis (see Chapter 59).1 Therefore, the terms acute pancreatitis and chronic pancreatitis describe syndromes with similar clinical and pathologic characteristics caused by multiple etiologies and varying mechanistic pathways.2
Hereditary pancreatitis refers to recurrent acute or chronic pancreatitis in an individual from a family in which the pancreatitis phenotype appears to be inherited through a disease-causing gene mutation expressed in an autosomal dominant pattern. Individuals with pancreatitis who carry a gene mutation that causes autosomal dominant pancreatitis have hereditary pancreatitis. Familial pancreatitis refers to pancreatitis from any cause that occurs in a family with an incidence that is greater than would be expected by chance alone, given the size of the family and incidence of pancreatitis within a defined population. Familial pancreatitis may or may not be caused by a genetic defect. Tropical pancreatitis (TP) is a form of early age-onset, nonalcoholic chronic pancreatitis occurring in tropical regions3 that is often clustered among family members, and that has a complex genetic basis. TP is further subdivided into fibrocalculous pancreatic diabetes (FCPD) and tropical calcific pancreatitis (TCP) based on the presenting feature of diabetes with fibrosis (FCPD) or severe pain with fibrosis and calcifications (TCP).4 The majority of children previously classified as having early onset idiopathic chronic pancreatitis have identifiable genetic etiologies, but there is no consensus terminology to distinguish the clinical diagnosis from the underlying etiology, especially when there is significant overlap.
MODELS OF PANCREATITIS AS A COMPLEX DISORDER
New models are necessary to understand the relationship among risk factors, etiologies, and the pathology of pancreatic disorders.5,6 Chronic pancreatitis, for example, can no longer be approached from an allopathic perspective (germ theory of disease in which one etiologic agent is responsible for a specific disease), but from the perspective of personalized medicine in which multiple factors must be considered in designing patient-specific treatments. For historical reasons most knowledge about chronic pancreatitis was derived from linking clinical symptoms in patients with abdominal pain and loss of pancreatic exocrine and endocrine function to pathologic changes in the pancreas observed at autopsy. The goal was to find the agent that caused chronic pancreatitis. Because of this approach the major advances in the late 20th century were technical in nature, allowing physicians to obtain biopsies (or high-resolution images) without an autopsies.7 Therapeutic interventions were directed at replacing lost function and attempting to control pain. However, the factors that determine which patient with known environmental risk factors (e.g., alcoholism and tobacco smoking) would progress to chronic pancreatitis whereas others did not, or why other patients without identifiable environmental risk factors progressed from a normal pancreas to chronic pancreatitis remained obscure. Resolving the multiple etiologies and mechanistic pathways could not be accomplished by examining end-stage pathology or by comparative gene expression profiles among patients with a normal pancreas and those with chronic pancreatitis because gene profiling defines abnormalities at a molecular level without discerning the proximal causes of the disease process.
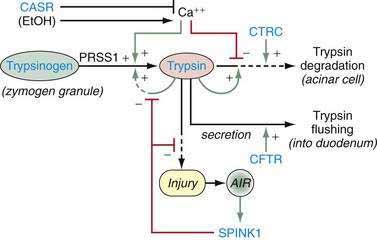
A new etiologic model of pancreatitis became necessary when it was discovered that the major genetic susceptibility factors for chronic pancreatitis actually caused acute pancreatitis through genetic defects that affected the ability of the pancreas to protect itself from premature trypsin activation.8 Trypsin was recognized as the critical molecule in pancreatitis because it is the master enzyme that controls activation of the other digestive enzymes inside the pancreas, and these enzymes cause tissue injury that triggers an inflammatory response. Most factors that increase susceptibility to pancreatitis disrupt a mechanism protecting the pancreas from trypsin-associated injury. Because trypsinogen is synthesized, stored, and transported almost exclusively by the pancreas, loss of mechanisms that protect the body from trypsin-associated injury specifically target the pancreas for recurrent injury. The classic example is hereditary chronic pancreatitis in which it has been observed that affected individuals experience recurrent acute pancreatitis that precedes chronic pancreatitis by a number of years.9–11 It is now recognized that a variety of genetic mutations exists that each increase the risk of pancreatitis through inadequate injury protection from trypsin. Furthermore, individuals with pancreatitis susceptibility genes only have occasional episodes of acute pancreatitis that are often triggered by identifiable environmental factors such as alcohol consumption or cigarette smoking. Because any number of stressors can trigger pancreatitis in genetically susceptible individuals, these factors can be categorized as metabolic and environmental stressors. Some individuals who inherit pancreatitis susceptibility genes and other genetically intact individuals exposed to strong environmental stimuli never develop recognizable acute pancreatitis, suggesting that triggering recurrent acute pancreatitis requires at least one factor in both domains.
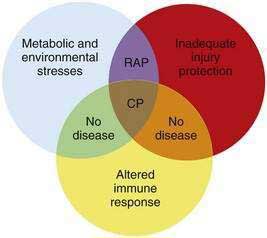
Another observation is that not all patients carrying the hereditary pancreatitis gene who have recurrent acute pancreatitis develop chronic pancreatitis.11 Chronic pancreatitis is characterized as chronic inflammation and fibrosis, which are immune-mediated processes.1 Fibrosis is the product of activated pancreatic stellate cells that are driven by anti-inflammatory cytokines including transforming growth factor-β (TGF-β) and interleukin-10 (IL-10).12–14 The inflammatory process is independent of the mechanism of injury and can be modified by genetic and environmental factors that influence the severity of fibrosis.8,15 If the normal response to pancreatic injury is recovery, then a complication of recurrent acute pancreatitis is extensive fibrosis (i.e., chronic pancreatitis). Factors that affect the immune system by accelerating fibrosis can be grouped together in the category of altered immune response to pancreatic injury. Figure 57-1 illustrates the complexity of chronic pancreatitis and the typical interaction of at least three different categories of risk factors before chronic pancreatitis develops. The study of pancreatic diseases, including chronic pancreatitis, must advance to predictive mathematical models that include all of the relevant risk factors and variables that are common in the human population, so that patient-specific treatments can be designed. This chapter focuses on the major genetic variables that will necessarily become part of the eventual predictive models, but that provide insight into specific disease syndromes that are already recognized.
MAJOR SUSCEPTIBILITY GENES CAUSING PANCREATIC DISEASE
CATIONIC TRYPSINOGEN GENE MUTATIONS
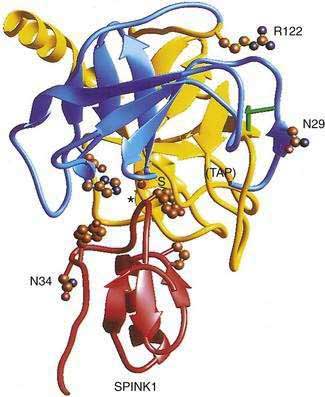
The cationic trypsinogen gene (UniGene name: protease, serine 1; PRSS1) was identified as the first pancreatitis-specific susceptibility gene through genetic linkage studies in families with hereditary pancreatitis.9 Cationic trypsinogen is the major form of trypsinogen (≈65%) followed by anionic trypsinogen (PRSS2, ≈30%), and mesotrypsin (PRSS3, ≈5%). The trypsin molecule is formed by a single peptide that folds into an enzyme with an active site between two globular domains linked by a single connecting chain. An eight amino acid extension of the enzyme, called the trypsinogen activation peptide (TAP), maintains the enzyme as inactive trypsinogen until it is cleaved by enterokinase or another trypsin molecule (autoactivation). Cleavage of TAP allows a conformation change that activates trypsin. Trypsin is also susceptible to trypsin-mediated autolysis beginning at the arginine 122 (R122) site of the connecting chain. The connecting chain is further degraded by chymotrypsin C (CTRC),16 which was biochemically characterized as enzyme Y.17 Finally, the trypsinogen molecule also has two separate calcium binding pockets that play key roles in trypsin regulation by exposing the activation site and blocking the autolysis site, respectively.
Trypsin plays a critical role in pancreatic physiology as the activator of the other pancreatic zymogens, a process that normally occurs within the duodenum where the zymogen activation cascade is initiated by the enterokinase-stimulated conversion of trypsinogen to trypsin. Trypsinogen activation and trypsin inactivation are primarily controlled by trypsin (autoactivation and autolysis), and the ambient calcium concentration serves as the switch between on and off (Fig. 57-2).8 Calcium binding to the first binding pocket that is formed by four aspartic acids within the TAP portion of trypsinogen facilitates trypsinogen activation by trypsin. Calcium binding to the second calcium binding pocket formed by a peptide loop in both trypsinogen and trypsin located adjacent to the autolysis loop, prevents exposure of the trypsin-sensitive R122 autolysis site and thereby prevents autolysis. Thus, physiologic regulation of trypsin activity is determined by cellular calcium, with increased cellular calcium facilitating activation and preventing inactivation and low cellular calcium levels limiting activation and permitting autolysis.
The maintenance of low calcium concentrations within the acinar cells is critical to protecting them from premature trypsinogen activation. However, other protective mechanisms are used to limit trypsinogen activation within the high calcium concentrations of the pancreatic duct fluid. Acinar cell calcium can rise through neurohormonal hyperstimulation (which opens basolateral calcium channels and is linked to calcium tunnels transporting calcium to the acinar pole)18,19; high extracellular calcium concentrations and submaximal pancreatic stimulation20; bile acid reflux, which opens apical membrane calcium pathways21; and prolonged, high-dose alcohol consumption, which lowers the threshold for stimulation-induced acute pancreatitis,22 possibly through mitochondrial damage23 and other factors that regulate intracellular calcium.24 Any process that increases acinar cell calcium will predispose to acute pancreatitis through a calcium-dependent trypsinogen activation and stabilization mechanism.24
More than 20 mutations have been identified in PRSS1 that increase susceptibility to recurrent acute pancreatitis,25 although the R122H and N29I mutations are most common. The locations of the R122H and N29I (N21I) mutations are shown in relationship to the active site in Figure 57-2 and in a mechanistic model in Figure 57-3. The mutations are clustered in regions associated with calcium-dependent trypsin regulation and may confer “gain-of-function” features by facilitating trypsinogen activation or retarding trypsinogen inactivation independent of cell calcium. Gain-of-function mutations often result in an autosomal dominant inheritance pattern; only one of the two trypsinogen alleles must code for a super-functional trypsin in order to prematurely trigger the zymogen activation cascade and cause pancreatitis, thus manifesting the phenotype. Other trypsinogen mutations that are unrelated to calcium-dependent trypsin regulation may predispose to recurrent pancreatitis by altering the activation or inactivation process normally regulated by pH or through interaction with other molecules,26 but the clinical relevance of these potential types of trypsinogen variants remains an area of investigation. The fact that the trypsinogen molecule has two calcium “switch” sites may explain why pancreatitis occurs only intermittently. The clinically important trypsinogen mutations and polymorphisms are discussed following.
The importance of maintaining a tight regulation of cationic trypsinogen is further highlighted by additional genetic findings. First, the R122H and N29I mutations appear to be gene conversions, in which a segment of deoxyribonucleic acid (DNA) from another similar gene replaces a similar segment of the gene of interest. There is now strong evidence that the “H” of PRSS1 R122H is a conversion mutation from Tryp 6,27 and the “I” of N29I is from PRSS2.28,29 These findings highlight the critical importance of the high-fidelity regulatory mechanism of PRSS1 in preventing recurrent, premature trypsinogen activation or trypsin survival in low-calcium environments. Secondly, it has been reported that gene copy number variants are also associated with a risk of chronic pancreatitis in France.30,31 In this case a segment of the genome containing the trypsinogen genes is duplicated on one chromosome, and when combined with the expression of trypsinogen for the opposite allele gives an extra “dose” of trypsinogen—enough to increase the risk of pancreatitis.
ANIONIC TRYPSINOGEN GENE MUTATIONS
Anionic trypsinogen (UniGene name: protease, serine 2; PRSS2) is a form of pancreatic trypsinogen that is usually expressed at about half the amount as cationic trypsinogen, although this ratio may change in some cases.32,33 To date, no gain-of-function mutations have been identified. However, a loss-of-function mutation, PRSS2 G191R, is associated with protection from pancreatitis.34,35 The mutation introduces an arginine (“R”) into a surface loop of PRSS2, making it a target for trypsin-mediated degradation.
CALCIUM-SENSING RECEPTOR GENE POLYMORPHISMS
Regulation of intra-acinar cell calcium is critical for the prevention of pancreatic injury.24,36 The calcium-sensing receptor (CASR) is a membrane-bound member of the G-protein–coupled receptor superfamily.37 CASR plays an important role in calcium homeostasis, as is reflected in its expression by cells of the parathyroid gland and renal tubules that are involved in the calcium metabolism. CASR has been identified in human pancreatic acinar and ductal cells, as well as in various nonexocrine tissues,38 although its functional significance in the pancreas has not yet been determined. More than 100 functional mutations (40 activating and 72 inactivating) have been described in the CASR mutation database related to familial hypocalcuric hypercalcemia (FHH), neonatal severe primary hyperparathyroidism (NSPHT), autosomal dominant hypocalcaemia (ADH) and related hypercalcemic or hypocalcemic disorders.39
In 2003 Felderbauer40 investigated a kindred with familial pancreatitis and a serine protease inhibitor Kazal type 1 (SPINK1) gene mutation (see following for discussion of SPINK1 mutations). However, only two of these family members had chronic pancreatitis, and both were found to have a novel CASR 518T>C mutation that was linked to hypercalcemia. An association between additional CASR variants, with or without SPINK1 mutations, was subsequently identified in patients in India with TP,41 as well as in the United States in sporadic and alcoholic chronic pancreatitis in which the CASR mutation doubles and triples the relative risk, respectively.42 The finding of different CASR polymorphisms in different populations is intriguing, but it appears that the presumed mild hypercalcemia is a cofactor rather than an independent risk factor, as seen in animal models of hypercalcemia.20,43 At the present time, CASR mutations analysis is not offered for clinical decision making.
CYSTIC FIBROSIS TRANSMEMBRANE CONDUCTANCE REGULATOR GENE MUTATIONS
The cystic fibrosis transmembrane conductance regulator (CFTR) gene is the most important molecule for regulating pancreatic duct cell function. This includes generating the bicarbonate-rich pancreatic juice that helps neutralize the acidic chyme coming from the stomach and flushing digestive enzymes out of the pancreas and into the duodenum. Both functions are accomplished by anion secretion through the CFTR located on the apical side of the pancreatic duct cells (see Chapter 56). The CFTR molecule becomes relevant to pancreatic diseases when its function, or regulation of its function, is altered by various gene mutations.
The CFTR molecule forms a regulated ion channel expressed on epithelial cells in the respiratory system, sweat glands, the digestive tract mucosa, biliary epithelium, pancreatic duct cells, and other locations. The primary anions conducted through CFTR under physiologic conditions are chloride and, under some conditions, bicarbonate. The CFTR gene contains more than 4300 nucleotides, with 24 exons and three splice variants that code for a single protein of 1480 amino acids.44 The CFTR molecule has 12 membrane spanning domains, 2 nucleotide-binding domains (NBD1 and NBD2), and a regulatory domain (R domain) with multiple phosphorylation sites (Fig. 57-4).
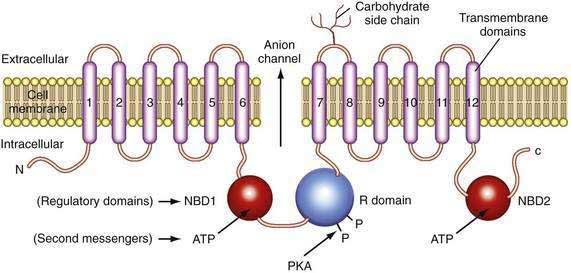
Although the regulation of CFTR is complex, many of the components that are relevant to the pancreas are now understood. CFTR-associated secretion is stimulated when the duct cell is stimulated by secretin or vasoactive intestinal peptide (VIP) acting on receptors that increase intracellular cyclic adenosine monophosphate (cAMP). The cAMP activates protein kinase A–mediated phosphorylation of various sites in the R domain, followed by increased anion conductance (e.g., chloride, bicarbonate) through the CFTR channel. The function associated with the individual phosphorylation sites on the R domain differs.45 Possible consequences of R domain phosphorylation include movement and insertion into the apical membrane, increased or decreased channel activity or specificity, or stabilization of other parts of the molecule such as NBD1. Duct cell stimulation by cholinergic agents or other agonists that increase intracellular calcium also potentiate anion secretion. Recent studies demonstrate that chloride conductance is regulated by cytoplasmic glutamate, while bicarbonate conductance is regulated by binding of ATP to NBD1 and NBD2,46 which may form a heterodimer to maximize adenosine triphosphatase (ATPase) activity.47 Structural studies of the CFTR protein suggest that the molecule exists in two different conformations depending on the presence or absence of ATP binding to NBD1 and NBD2.48 If the basolateral membrane of the duct cell is nearly impermeable to chloride during bicarbonate secretion and if bicarbonate conductance through CFTR is limited because of an unfavorable CFTR conformation, then net ion transport across the basolateral and apical membranes would markedly be reduced49 and the duct cells would not be able to help flush digestive enzymes out of the duct. This would put the pancreas at risk of recurrent acute pancreatitis because prematurely activated digestive enzymes would not be removed from the pancreas.
Major mutations in both CFTR alleles result in loss of CFTR function. The consequences include inability to adequately hydrate mucus and other macromolecules, leading to accumulation of viscid material and inspissated glands. This condition results in progressive organ destruction of the pancreas and respiratory system, and dysfunction of the liver, intestine, sweat glands, and other sites where epithelial cell secretion plays an important physiologic role. The pancreas incurs a double risk because much of its proteins are zymogens and trypsin activation will lead to recurrent injury and eventually destruction of the pancreas through progressive fibrosis. Trypsin-mediated injury and destruction of the pancreas in children with CF is consistent with this model because the pancreatic pathology in CF is pseudocyst formation and fibrosis rather than atrophy (as expected with duct obstruction).50 It appears that pancreatic gland injury in CF children roughly parallels the expression of trypsinogen in the developing acinar cells, which begins at 16 weeks’ gestation and gradually increases in concentration until birth and through the first six months of life when levels markedly rise.51,52 The resulting histology has many of the features of end-stage chronic pancreatitis that develops in children and adults, but also has striking expanded ducts that appear as multiple protein filled cysts (Fig. 57-5).
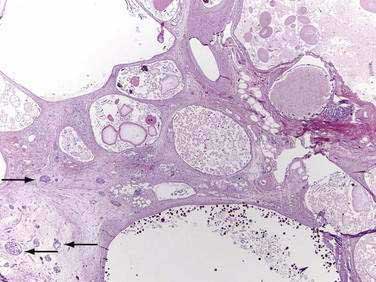
The overall clinical picture in an individual case depends on the nature of the combined CFTR mutations, the genetic background in which the defective genes operates (e.g., modifier genes), and environmental factors.50,53 About 70% of white patients with CF have a three-base pair deletion of the phenylalanine-coding codon 508 (?F508), although 1600 other mutations have been reported. Distinct mutations are common to certain ethnic groups, including African Americans who carry the 3120+1G>A mutation at a frequency of 12.3%54 and the R334W mutation in Hispanics, which is associated with CF with pancreatic sufficiency (PS) but recurrent acute pancreatitis.55 Patients with one severe CFTR mutation and one mild CFTR mutation (e.g., R117H or R334W) often have CF with pancreatic sufficiency.56,57 The reason may be that mutations such as R117H markedly reduce chloride conductance without affecting bicarbonate secretion.46,58 Reduction of chloride conductance would affect all of the epithelial cells in organs that use CFTR to transport chloride, but would have much less effect on the pancreatic duct because it uses CFTR to transport bicarbonate.49 On the other hand, CFTR mutations that specifically inhibit bicarbonate secretion put the pancreas at risk of recurrent pancreatitis without affecting the organs that use CFTR to transport chloride.
The functional consequence of CFTR mutations depends on the combined effects of both CFTR alleles with the severity of the phenotype dependent on the mildest mutation.59 The most common CFTR mutations have been organized according to the effect on clinical phenotype (severe, mild-variable, borderline, benign) and the effect on CFTR protein structure and function (classes 1 to 5).50,53 (Examples are given in Table 57-1.) Class 1 to 3 mutations result in no functional protein and therefore are associated with a severe phenotype when combined with a second severe CFTR mutation. Class 4 and 5 CFTR mutations result in CFTR proteins with altered, but residual function, and are associated with mild-variable or borderline phenotypes. There is current debate as to whether additional classes are justified. If a class 4 or 5 mutation associates with a class 1 to 3 mutation, the phenotype is mild CF, with only a subset of organs affected.60 The resulting conditions are often called atypical CF.59 If a class 1 to 3 or some class 4 mutations are combined with wild-type CFTR or a benign polymorphism, the overall function of CFTR is reduced by up to 50%, but the phenotype is usually normal because more than 90% of overall CFTR function must be lost before clinical features of atypical CF are seen.59 Recurrent acute pancreatitis requires, at minimum, a partially functioning pancreas and is therefore seen in some cases of CF with pancreatic sufficiency and atypical CF.
Table 57-1 Classification of CFTR Mutations, Resulting Defect, and Associated Degree of Pancreatic Dysfunction

Many of the features of CF cannot be explained by variations in CFTR sequence. Instead, these features are caused by specific environmental factors or modifier genes.50,61 Environmental factors, such as bacterial colonization of the respiratory system, tobacco smoke,62 and poor nutritional status contribute to the severity of lung disease.63 The risk of liver disease appears to be independent of CFTR genotype; it is associated with meconium ileus.64 Careful consideration must be given to patient with either classic CF symptoms, or atypical presentations resulting from less common combinations of genetic and environmental factors.
In 1998 two groups65,66 demonstrated that CFTR mutations were also very common in idiopathic and alcoholic chronic pancreatitis, which indicates that some of the more than 1600 known CFTR gene sequence variants cause milder disease (atypical CF59), pancreas-specific injury or that CFTR gene mutations may be part of a more complex trait.1 In some cases, recurrent acute pancreatitis and chronic pancreatitis appeared to be associated with heterozygous CFTR genotypes. Because heterozygous CFTR mutations and polymorphisms are common in the general European and American population, and because the parents of CF children (obligate CFTR mutation carriers without CF) do not have an increased incidence of acute or chronic pancreatitis compared with the normal population,67 it is likely that a second factor that specifically targets the pancreas is required.1 In early onset idiopathic pancreatitis this second factor may be a SPINK1 mutation (discussed later), a potent environmental factor,68 or other complex genetic combinations.69–73
SHWACHMAN-BODIAN-DIAMOND GENE MUTATIONS
The Shwachman-Bodian-Diamond syndrome gene (SBDS) is a gene of unknown function that is mutated in most cases of Shwachman-Diamond syndrome (SDS).74,75 This gene has five exons and encodes a predicted protein of 250 amino acids. The genetic defect in most cases of SDS is caused by gene conversion between the normal SBDS gene with a nonfunctional pseudogene designated SBDSP.74 The SBDSP pseudogene DNA code is 97% identical to SBDS gene code. The differences between the SBDS gene and SBDSP pseudogene are critical nucleotide deletions and nucleotide changes that render the SBDS gene product nonfunctional. Fourteen distinct mutations were initially identified in these kindreds, with the most common being the conversion mutations 183-184TA→CT and 258+2T→C. Interestingly, most patients had compound heterozygous mutations, and no patient was homozygous for the common 183-184TA→CT mutation. Although the function of the gene is unknown, there is significant homology with genes in other species that regulate or facilitate mRNA utilization or metabolism.74 Recent studies suggest that the gene encodes a protein that interacts with ribosomal RNA and helps regulate the maturation of the 60s ribosomal subunit and may also stabilize the mitotic spindle.76–78 The genetic defect results in an acinar cell–specific defect with markedly reduced zymogen synthesis and pancreatic insufficiency rather than susceptibility to pancreatitis. The other clinical features of the SDS are discussed later.
MODIFIER GENE
PANCREATIC SECRETORY TRYPSIN INHIBITOR GENE MUTATIONS
Pancreatic secretory trypsin inhibitor (PSTI, UniGene name: serine protease inhibitor, Kazal type 1; SPINK1) is a 56 amino acid peptide that specifically inhibits trypsin by physically blocking its active site (see Fig. 57-2). SPINK1 is synthesized by pancreatic acinar cells along with trypsinogen and it co-localizes with trypsinogen in the zymogen granules. In the mechanistic models of pancreatic acinar cell protection, SPINK1 acts as the first line of defense against prematurely activated trypsinogen in the acinar cell.9,32,79 SPINK1 is an acute phase reactant and concentrations in serum rise markedly with inflammation.80,81 Under normal conditions there is a great excess of potential trypsin compared with SPINK1 so that the inhibitory capacity of SPINK1 is limited, but with inflammation the expression of SPINK1 is increased dramatically.82
A number of SPINK1 mutations have been identified, but a high-risk haplotype (five polymorphisms that are inherited together) defined by SPINK1 N34S is by far the most common, being present in 1% to 4% of most populations throughout the world.79,83,84 Several other variants of the SPINK1 gene also have been described.25 Pancreatitis associated with SPINK1 gene mutations is associated with early onset recurrent acute and chronic pancreatitis in children,79 familial pancreatitis,83 and TP,85 and is often a feature of polygenic pancreatitis–associated genotype.
Because SPINK1 is a specific trypsin inhibitor, and because expression of SPINK1 is normally very low,82 it follows that SPINK1 cannot be a significant inhibitor of prematurely activated trypsin until the acute inflammatory process has become well established. Thus, the effect of loss-of-function SPINK1 polymorphisms would only become apparent late in the course of acute pancreatitis, or in the case of recurrent premature trypsinogen activation, as is seen with PRSS1 or CFTR mutations. This hypothesis was tested using multiple meta-analyses on data that were classified by proximal etiology.86 This analysis suggested that the strongest effect of SPINK1 N34S was in pancreatitis etiologies that were linked with recurrent trypsin activation, but the effect was low in other etiologies (e.g., alcohol-associated pancreatitis). If this is mechanistically true, then up-regulation of mutant SPINK1 will fail to prevent trypsin-associated recurrent pancreatic injury. Furthermore, suppression of recurrent proinflammatory responses with macrophage-derived anti-inflammatory cytokines (e.g., TGF-β1) will drive the pancreatic stellate cells to produce fibrosis.7,14,87 The implication is that the discovery of a SPINK1 mutation in an unaffected individual is of minimal importance, whereas the effect of a SPINK1 mutation in driving fibrosis in someone with PRSS1 or CFTR mutations is very strong. A summary of the mechanisms for trypsin activation and inactivation is illustrated in Figure 57-3.
PANCREATIC DISEASE IN CHILDREN
Whereas pancreatic disease in children was once considered uncommon, evidence suggests that the incidence is increasing. Although diagnostic modalities and physician awareness continue to improve, this does not appear to account for the increase.88 Acute pancreatitis occurs in all age groups, including infants.89 The most common causes of acute pancreatitis in adults are excessive alcohol use and gallstones. These risk factors are less often seen in children, although biliary pancreatitis is now recognized in this age group. The majority of cases of recurrent acute and chronic pancreatitis in children have a structural or genetic basis. The genetic factors predisposing to acute pancreatitis appear to be similar to those associated with chronic pancreatitis and are discussed in detail in the following sections.
ETIOLOGY OF ACUTE PANCREATITIS
The primary causes of acquired pancreatitis in children are listed in Table 57-2. In a recent review of 1276 children with acute pancreatitis compiled from five studies, the most common causes were idiopathic (22.2%), association with systemic disease (20.8%), trauma (18.6%), structural (e.g., pancreas divisum) (10.6%), and medications (10.2%); gallstones, post–endoscopic retrograde cholangiopancreatography (ERCP), familial, hypercalcemia, hyperlipidemia, diabetic ketoacidosis, and “other” causes made up the remaining etiologies.88 Growing evidence suggests that some of these cases occur in children with high-risk genetic alterations, especially pancreatic-specific combinations of SPINK1 and CFTR mutations. Genetic testing, discussed later in this chapter, is usually performed after recurrent episodes and when other common causes have been excluded.
Table 57-2 Reported Causes of Acquired Pancreatitis in Children
AIDS, acquired immunodeficiency syndrome; ERCP, endoscopic retrograde cholangiopancreatography; HIV, human immunodeficiency virus.
* Class 1 drug: pancreatitis occurs with rechallenge(see Chapter 58).
† Probable cause of pancreatitis.
Trauma
Trauma is a frequent cause of acute pancreatitis despite the fact that the pancreas is well protected from minor injury by its retroperitoneal location. The trauma is usually blunt, associated with injuries to other abdominal viscera and becomes evident soon after the injury,90 although injury may apparently precede the manifestation or recognition of pancreatitis by several weeks. In such an instance, a precise relationship is unclear. Perhaps of more importance is that the possibility of injury to the pancreas is often not considered in a severely injured or battered child.91
Structural Abnormalities
Structural abnormalities are being recognized earlier as imaging techniques such as magnetic resonance imaging (MRI) and magnetic resonance cholangiopancreatography (MRCP) improve. Pancreas divisum is the most common anatomic aberration, although a wide variety of other structural abnormalities of the bile and pancreatic duct also have been observed (see Chapter 55).88
The widespread availability of MRCP has drastically reduced the use of diagnostic ERCP. Post-ERCP pancreatitis has been a significant cause of pancreatitis in several series,92,93 and this etiology is seen wherever ERCP is performed in children. ERCP remains invaluable for therapeutic intervention (see Chapter 61).
Biliary Tract Disease
Gallstone pancreatitis is less common in children than in adults and is probably a reflection of the relative infrequency of cholelithiasis before puberty. However, because almost 10% (as high as 30% in a single study) of children with pancreatitis in some series had cystic duct stones or common bile duct disease, this diagnosis must be considered, regardless of age.94 Little is known about the natural history of this disease in children.
Medications
Medications remain a frequent cause of acute pancreatitis in children, although the disease underlying the prescription must also be considered in the differential diagnosis.95–97 Recent studies identified valproate as the most frequent drug associated with pancreatitis in children, followed by L-asparaginase, prednisone, or multiple medications.92,93,98 The development of persistent abdominal pain in a child receiving any medication should suggest the possibility of drug-induced pancreatitis. This is confirmed only by documentation of pancreatic disease, improvement on drug withdrawal, and return of disease when the drug is reintroduced.
Infection
Infections, particularly with viruses, are a frequently associated with childhood pancreatitis; a partial list of putative agents appears in Table 57-2. Enteroviruses, particularly coxsackievirus and echovirus, have been documented by stool isolation and concomitant serum titer rise in up to 8% of adults with idiopathic acute pancreatitis. Only about half of virus isolations are associated with an antibody rise.99–101 Pancreatitis has been reported in children with Epstein-Barr virus infections, often appearing after an initial clinical improvement.102,103 Interstitial pancreatitis has been described in the congenital rubella syndrome.104 Pancreatitis in children is often attributed to mumps virus on the basis of abdominal pain and an elevated serum amylase value, with parotitis or waxing mumps antibody titers or both.105 Confirmation by serum isoamylase or lipase determinations and by abdominal ultrasonography is lacking, however, and the frequency of this entity may be overestimated. Bacterial pancreatitis has been described in one patient,106 although in that patient there may have been other causes for development of pancreatitis, including antecedent hypotension. Mycoplasma pneumoniae infection, followed one to two weeks later by clinically apparent pancreatitis, has been seen in an estimated 8% of patients with this infection. Complement-fixation titers and serum immunoglobulin M values were elevated, and other causes of pancreatitis were absent.107 Typhoid fever often manifests with abdominal pain; pancreatitis has been suggested as one possible cause.108 Although uncommon in the United States, ascariasis is among the most frequent causes of pancreatitis in children in regions such as South Africa and India. Worms can be found within the pancreatic duct and can be vomited as the initial diagnostic clue. Malaria also has been reported to cause pancreatitis.109 Pancreatitis is 35 to 800 times more common in patients with acquired immunodeficiency syndrome (AIDS).110 This extremely high risk for acute pancreatitis is attributed to several factors (see Chapter 33). A number of medications that are frequently prescribed to human immunodeficiency virus (HIV)–infected patients are associated with pancreatitis (see Table 57-2), possibly due to direct toxicity to pancreatic acinar cells. In addition, immunodeficiency itself predisposes patients to pancreatic infection.
Acute Pancreatitis in Systemic Diseases
Acute pancreatitis is often seen in patients with severe systemic illnesses.88,93 Hemolytic uremic syndrome (HUS) was the most common cause of acute pancreatitis of all systemic diseases in two major studies.92,93 The mechanism is unknown and likely multifactorial, and uremia itself is a risk factor for pancreatic injury.111–113 Systemic lupus erythematosus has been reported in association with pancreatitis.90
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


