CHAPTER 78 Hepatitis B and D
HEPATITIS B
An estimated 400 million persons are carriers of hepatitis B virus (HBV) in the world today; of these, 75% reside in Asia and the Western Pacific. Effective vaccines against HBV have been available since the early 1980s, but perinatal and early life exposures continue to be major sources of infection in high-prevalence areas. High-risk behaviors such as promiscuous heterosexual contact and injection drug use account for many new cases in young adults. Fulminant acute hepatitis B accounts for several hundred deaths per year in the United States, and chronic HBV infection accounts for one million deaths worldwide each year from complications of end-stage liver disease, including hepatocellular carcinoma (HCC). Hepatitis B is the chief cause of cirrhosis and HCC in the world today, and nationwide vaccination has been shown to diminish greatly the number of new cases of infection and HCC in Taiwanese children.1 Universal hepatitis B vaccination is likely to have the greatest impact on liver disease–related mortality in future generations.
EPIDEMIOLOGY
Geographic Distribution and Sources of Exposure
The prevalence of hepatitis B varies markedly around the world. In highly endemic regions, such as Southeast Asia (excluding Japan), China, and much of Africa, 8% or more of the population are chronic HBV carriers, and the lifetime risk of infection ranges from 60% to 80%.2 In these areas, perinatal transmission and horizontal spread among children are the major sources of infection. Approximately 60% of the world’s population reside in these areas where HBV is highly endemic.3 Regions of intermediate risk include parts of southern and eastern Europe, the Middle East, Japan, the Indian subcontinent, much of the former Soviet Union, and northern Africa. In intermediate-risk areas, the lifetime risk of infection is between 20% and 60%. Persons of all age groups are infected, but as in high-risk areas, most infections occur during infancy or early childhood. Regions of low prevalence are North America, western Europe, certain parts of South America, and Australia. In these areas, the lifetime risk of HBV infection is less than 20%, and transmission is primarily horizontal (i.e., between young adults). Sexual transmission is the main mode of transmission in Europe and North America, and injection drug use is a major contributor to new cases as well.4
Transmission of infection from an HBV carrier mother to her neonate accounts for the majority of new infections in the world today. Sixty to ninety percent of hepatitis B surface antigen (HBsAg)-positive mothers who are hepatitis B e antigen (HBeAg)-positive transmit the disease to their offspring, whereas mothers who are positive for antibody to HBeAg (anti-HBe) transmit the disease less frequently (15% to 20%) (see later discussion of diagnosis). Other less common sources of infection are household contact with an HBV carrier, hemodialysis, exposure to infected health care workers, tattooing, body piercing, artificial insemination, and receipt of blood products or organs. Since routine screening of the blood supply was implemented in the early 1970s, transfusion-associated hepatitis B has become rare in the United States. Hepatitis B can be transmitted by blood that tests negative for HBsAg but positive for antibody to hepatitis B core antigen (anti-HBc) because of low levels of circulating HBV DNA in such blood.5 HBsAg-negative blood that is positive for anti-HBc is excluded from the donor pool in the United States and many countries around the world. In 0% to 30% of persons who are seropositive for anti-HBc alone, HBV DNA is detectable in serum by polymerase chain reaction (PCR) testing.6
HBV is transmitted efficiently by percutaneous and mucous membrane exposure to infectious body fluids. The virus is 100 times as infectious as human immunodeficiency virus (HIV) and 10 times as infectious as hepatitis C virus (HCV). HBeAg seropositivity indicates a higher risk of transmission from mother to child, after needlestick exposure, and in the setting of household contact. HBV DNA has been detected by sensitive techniques such as PCR testing in most body fluids, except for stool that has not been contaminated with blood. Although HBV replicates primarily in hepatocytes, the presence of replicative intermediates and virally encoded proteins in other sites, such as the adrenal gland, testis, colon, nerve ganglia, and skin, suggests that a vast extrahepatic reservoir for infectious virus exists.7 Small amounts of HBV DNA have been demonstrated in peripheral mononuclear cells and liver tissue years after apparent resolution of chronic infection.8,9 Extrahepatic localization of low levels of replicating virus explains the relatively high rate of transmission of infection from anti-HBc–positive organ donors.10
Rates of Infection in the United States
The incidence of hepatitis B has been declining in the United States over the past decade because of universal vaccination of newborns, adult vaccination programs for high risk persons, changes in sexual lifestyle, refinements in blood screening procedures, and the availability of virus-inactivated blood components.11 Most striking has been the decrease among children and health care workers, groups with the highest rates of vaccination. Data from the Centers for Disease Control and Prevention (CDC) indicate that more than 95% of pregnant women in the United States are tested for HBsAg, and infant vaccine coverage levels are now equivalent to those of other vaccines in the childhood schedule. Nonetheless, an estimated 78,000 new HBV infections occurred in 2001, with the highest incidence rates among sexually active young adults (20 to 29 years old) and higher rates occurring among black and Hispanic persons than in white persons.12 Since 1995, approximately 40% of cases of acute hepatitis B reported to the CDC were caused by intimate contact among heterosexuals, 15% to 20% were related to intravenous drug use, and 12% occurred in men who have sex with men. No identifiable source of exposure was demonstrated in approximately 15% of cases. Nearly one third of prison inmates have been infected with hepatitis B, and 2% are chronically infected.
According to the third National Health and Nutrition Examination Survey (1988 to 1994), one or more serologic markers of HBV infection were demonstrated in 4.9% of the U.S. population, and the prevalence of chronic infection was 0.2%.13 Traditional estimates based on the results of blood donation screening in the late 1970s also indicated a prevalence rate for chronic infection of 0.2% to 0.4% in the United States. Although traditional estimates are that the number of HBV carriers in the United States is between 1.25 and 1.5 million, this figure is likely to be a serious underestimate because of changing immigration patterns and underrepresentation of certain minority groups in field surveys. For example, 15 million Asians live in the United States, and even a conservative estimate that the prevalence in this group is 5% would raise the overall number of HBV carriers in the United States by more than 750,000.
CLINICAL OUTCOMES
Definitions
In common usage, the term HBV carrier has often been used to refer to persons persistently infected with HBV who have normal serum aminotransferase levels. (They are sometimes inappropriately referred to as “healthy” HBV carriers.) Because the nomenclature is potentially confusing, the proposal has been made that the carrier state be categorized as inactive or active. Inactive carriers are patients who have evidence of HBV replication on a PCR-based assay only (but not with a less sensitive non–PCR-based assay) and normal or only mildly elevated serum aminotransferase values (see later).14 Long-term follow-up of inactive carriers suggests that the majority of these patients do not have progressive liver disease and do not experience complications. Some of these patients, however, ultimately have one or more episodes of reactivated hepatitis in which the levels of viremia and serum aminotransferase activity increase. Also, some patients with the inactive carrier state may demonstrate HCC. Active carriers, on the other hand, have evidence of HBV replication on non–PCR-based assays for HBV DNA, intermittently or persistently elevated serum aminotransferase levels, and evidence of chronic hepatitis in a liver biopsy specimen.
Clinical Sequelae of Acute Hepatitis B Virus Infection
The age at which a person becomes infected with HBV is a principal determinant of the clinical outcome. HBV infection in adults with an intact immune system is likely to cause clinically apparent acute hepatitis B; only 1% to 5% of these persons become chronically infected.4 By contrast, as many as 95% of infected neonates become chronic HBV carriers because of immunologic tolerance to the virus.
In adults, fulminant liver failure caused by acute hepatitis B occurs in less than 1% of cases, but this group still accounts for 5% of all cases of acute liver failure and approximately 400 deaths annually in the United States.15 Rapid viral elimination may result in clearance of HBsAg from serum by the time of initial presentation. In these cases, the accurate diagnosis of fulminant hepatitis B may require testing with immunoglobulin (Ig) M antibody to hepatitis B core antigen (HBcAg) (IgM anti-HBc) (see later discussion of serologic markers of infection).16 The rate of spontaneous survival in acute liver failure caused by hepatitis B is only approximately 20%. Liver transplantation has resulted in survival rates of 50% to 60%. Recurrent disease in the allograft is now uncommon because of administration of hepatitis B immune globulin (HBIG) and potent orally administered antiviral agents (see later and Chapters 93 and 95).
Clinical Sequelae of Chronic Hepatitis B Virus Infection
The presence of active viral replication and long-standing necroinflammatory liver disease caused by HBV strongly influences the rate of progression to cirrhosis. The major determinant of survival is the severity of the liver disease when the patient first comes to medical attention.17 Cirrhosis is associated with decreased survival and an increased frequency of HCC. Five- and 20-year survival rates of 55% and 25%, respectively, have been reported in patients with cirrhosis at presentation, whereas rates of 97% and 63%, respectively, have been reported for those with mild (noncirrhotic) disease.18 Survival rates differ most dramatically between patients with compensated and decompensated cirrhosis. In one study, an 84% five-year survival rate was reported for patients with compensated HBV-related cirrhosis, compared with 14% for patients with cirrhosis complicated by ascites, jaundice, encephalopathy, or a history of variceal bleeding.19 Multivariate analyses in several large cohort studies have identified age, ascites, hyperbilirubinemia, and other features of advanced liver disease as correlating independently with survival in patients with HBV-related cirrhosis. Interferon-induced clearance of HBeAg (see later) has been associated with prolongation of survival without complications or the need for liver transplantation.20
Clearance of HBsAg in patients with HBV-related cirrhosis has been associated with an excellent prognosis, including improvement in liver histology and function, a decreased chance of viral reactivation, and prolonged survival.17 Even HBsAg clearance, however, is not an absolute safeguard against the future development of HCC in persons who already have cirrhosis.21
MOLECULAR BIOLOGY
HBV is a small DNA virus that belongs to the Hepadnaviridae family. Other members of this virus family are human HBV-like agents that infect the woodchuck, ground and tree squirrels, woolly monkey, crane, heron, Ross goose, and duck. HBV is a small (3.2-kilobase [kb]) virus with a DNA genome that has a relaxed, circular, partially double-stranded configuration (Fig. 78-1). The genome is composed of four open reading frames (ORFs) and has a compact design in which several genes overlap and use the same DNA to encode different viral proteins. The four viral genes are the core, surface, X, and polymerase genes. The core gene encodes the core nucleocapsid protein, which is important in viral packaging and production of HBeAg. The surface gene encodes the pre-S1, pre-S2, and S proteins (comprising the large [L], middle [M], and small [S] surface proteins). The X gene encodes the X protein, which has transactivating properties and may be important in hepatic carcinogenesis. The polymerase gene has a large ORF (approximately 800 amino acids) and overlaps the entire length of the surface ORF. It encodes a large protein with functions that are critical for packaging and DNA replication (including priming, RNA- and DNA-dependent DNA polymerase, and RNase H activities).
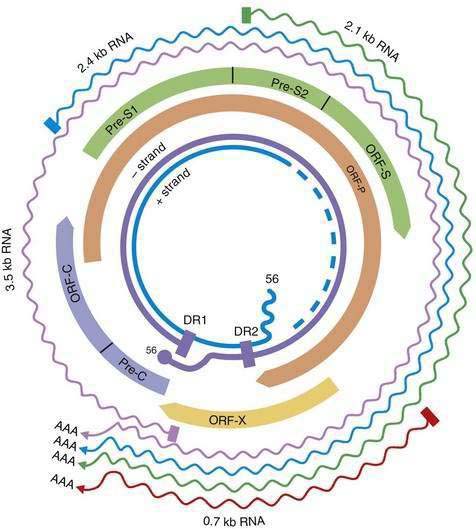
Although HBV is a DNA virus, replication occurs through an RNA intermediate and requires an active viral reverse transcriptase/polymerase enzyme. The mutation rate is higher for HBV than for other DNA viruses (an estimated 1010 to 1011 point mutations per day).22 Complete HBV genomic sequencing has identified a large number of mutations within the HBV genome, many of which are silent or do not alter the amino acid sequence of encoded proteins. Because of genomic overlap, however, some of the silent mutations in one ORF (for example, the polymerase gene) may result in an amino acid substitution in an overlapping ORF (surface gene), although with currently uncertain clinical implications.
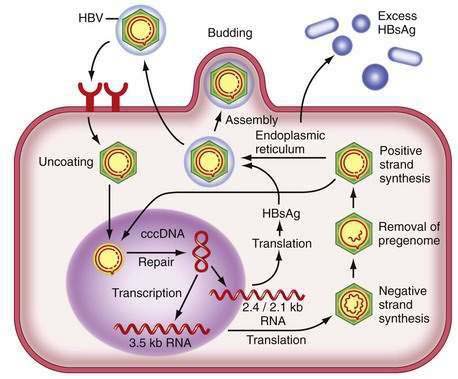
Figure 78-2 illustrates the life cycle of HBV. The initial phase of hepadnaviral infection involves the attachment of mature virions to host cell membranes. The human receptor for HBV remains unknown. Entry of the virus results from fusion of the viral and host membranes as the nucleocapsid is released into the cytoplasm. Mechanisms of intracellular transport of viral genome into the nucleus are poorly understood, but the first step in genomic replication involves conversion of the relaxed circular form of HBV DNA into a double-stranded, covalently closed circular form (cccDNA). The cccDNA, which serves as the template for viral transcription, is the major form of viral DNA in the nucleus of infected hepatocytes. Subgenomic (0.7 to 2.4 kb) and pregenomic (3.5 kb) RNA molecules are transcribed from this template. The L protein is translated from the 2.4 kb RNA, the M and S proteins from the 2.1 kb RNA, and the X protein from the 0.7 kb transcript. The pregenomic RNA serves as the template for reverse transcription as well as the messenger RNA (mRNA) for translation of the core and polymerase proteins; the precore RNA codes for the precore gene product.
Hepatitis B Virus Genotypes
A genetic classification based on comparisons of complete genomes has demonstrated eight genotypes of HBV, designated A through H (Table 78-1).23 Several methods have been used for HBV genotyping, including a commercially available line probe assay. Genotypic differences are based on an intergroup divergence of 8% or more in the complete nucleotide sequence. Genotype A is the predominant genotype in northern Europe and the United States. Genotypes B and C are confined to populations in eastern Asia and the Far East, but changes in immigration patterns have resulted in an influx of Asian HBV carriers with these genotypes into the United States.24 Genotype D is found worldwide but is especially prevalent in the Mediterranean area, Middle East, and south Asia. Genotype E is indigenous to western sub-Saharan areas, and genotype F prevails in Central America. Cases of genotype G have been reported in the United States and France. Genotype H has been described in Mexico.
Table 78-1 Hepatitis B Genotypes (A-H) and Their Possible Clinical Associations
| Geographic Distributions |
| Proposed Clinical Associations |
HBeAg, hepatitis B e antigen; HBsAg, hepatitis B surface antigen; HBV, hepatitis B virus.
Clinical associations appear to exist with the various genotypes (see Table 78-1). Currently, the strongest clinical associations appear to be that (1) HBeAg seroconversion occurs earlier in patients with genotype B than in those with genotype C and (2) response to therapy with interferon varies with genotype (see later).25 The viral genotype also has implications for the frequency of precore and core mutations (see later) and may have an effect on the frequency of HCC.
Mutations of the Hepatitis B Virus Genome
Hepatitis B Surface Antigen Mutants
Mutations in the surface gene can result in changes in the antibody-binding domain. Accordingly, both virus neutralization by polyclonal antibody to HBsAg and testing for HBsAg by methods that depend on antibody binding can be affected. Large-scale vaccination programs in regions endemic for HBV have revealed a 2% to 3% frequency of vaccine escape mutants that result from alterations in the “a” determinant of the HBsAg protein, which is the major neutralizable epitope. The typical mutation results in the substitution of glycine for arginine at amino acid position 145; this substitution prevents binding of neutralizing antibodies (i.e., antibody to HBsAg [anti-HBs]). The clinical significance of these mutants for neonatal vaccination programs is highly controversial because the frequency of these variants among HBV-infected mothers whose infants respond to vaccination has been found to be similar to that of mothers whose infants do not respond. The “a” determinant mutants also are proposed to have clinical relevance after liver transplantation for hepatitis B. As many as 50% of patients in whom recurrent HBV infection develops despite the use of HBIG have been shown to have these escape mutants, and the rate at which the mutations are detected appears to correlate with the length of time over which HBIG is repeatedly administered.26
Mutations in the Precore, Basal Core Promoter, and Core Genes
Mutations in the precore and basal core promoter regions of the HBV genome can influence the production of HBeAg. A precore mutation results in a stop codon at nucleotide 1896 that abolishes the synthesis of HBeAg,27 whereas mutations in the basal core promoter at nucleotides 1762 and 1764 decrease HBeAg synthesis by approximately 70% while maintaining pregenomic RNA levels.28 Both types of mutations have been observed in cases of severe or fulminant hepatitis, which has been attributed to the loss of the immune-tolerizing effects of HBeAg antigen (see later). The presence of core promoter mutations has been linked to a significantly increased risk of HCC.29 Precore and basal core promoter mutants have been described in the same patients and are particularly common in Asian and European patients with chronic hepatitis B.30 A large serosurvey of HBV carriers residing in the United States has found that precore and core promoter mutations are common (frequencies of 27% and 44%, respectively), depending on the ethnicity and places of birth of the patients. Both mutant forms of HBV were observed to occur far more commonly in HBeAg-negative patients (precore mutation in 38% of HBeAg-negative versus 9% of HBeAg-positive patients; core promoter mutation in 51% versus 36%).31 In addition to these mutations, upstream mutations in the core gene can influence immunologic responses to HBV. Core gene mutations have been shown to block recognition of HBV by cytotoxic T lymphocytes (CTLs), a key mode of viral clearance. Therefore, the mutations contribute to HBV immune escape and possibly influence the response to interferon.32 Core gene mutations within the immunodominant epitopes of the HBV nucleocapsid also can affect CD4+ T-cell reactivity.33
Hepatitis B Virus DNA Polymerase Mutants
The polymerase gene encodes a DNA polymerase enzyme needed for encapsidation of viral RNA into core particles, conversion of the pregenomic viral RNA into a negative strand of viral DNA (reverse transcription), and conversion of this first HBV DNA strand into a second DNA strand of positive polarity. In general, the HBV reverse transcriptase function of the polymerase gene is highly conserved because major mutations that impair the efficiency of viral replication lead to selection pressure against such variant forms. As indicated earlier, HBV has a high rate of replication (1011 virions per day) and low replication fidelity, meaning that it has a propensity to mispair nucleotide bases when it reverse transcribes viral RNA to DNA. HBV DNA polymerase also lacks any proofreading activity, so it cannot repair its mistakes. Therefore, when a nucleotide base is misplaced, it remains in the growing viral DNA strand as a base mutation, and the new HBV DNA molecule has a different sequence from the original (wild-type) genome. The overall error rate of HBV DNA polymerase is estimated to be 1 per 10,000 nucleotides copied, which translates to the potential for 10 million base-pair errors per day in an infected person. All possible single-base mutations can be produced in a 24-hour period, although many such mutations will yield nonviable viruses.34
Mutations in the sequence of HBV polymerase can lead to drug resistance to nucleoside analogs used to treat HBV infection because some HBV polymerase mutants have decreased susceptibility to these drugs and are selected during treatment. The mutations in the sequence of HBV DNA polymerase that confer drug resistance result in amino acid substitutions in the reverse transcriptase domain of the enzyme. The changes in the structure of the enzyme, in turn, are thought to sterically inhibit binding of the drugs to their active sites. The amino acids in the reverse transcriptase are numbered from 1 to 344, and an amino acid identity is given by the single letter amino acid code. By convention, substitutions in reverse transcriptase are designated by the wild-type amino acid, followed by the number of the amino acid, followed by the substituted amino acid. For example, for the nucleoside analog lamivudine (see later), two types of mutations occur at nucleotide position 204 of domain C (the catalytic site of the polymerase) that result in substitution of the amino acid methionine (M) for either isoleucine (I) or valine (V). These mutations are designated M204I and M204V, respectively, and are referred to collectively as YMDD mutants; the letters stand for the amino acids (Y = tyrosine, M = methionine, D = aspartate) in the C domain. The M204V mutation tends to occur in conjunction with a mutation in domain B that results in substitution of leucine (L) with methionine (L180M). The M204I mutation or the combined M204V-L180M mutations result in marked resistance to the effect of lamivudine (>10,000-fold reduction in susceptibility). After prolonged exposure to adefovir, drug resistant mutations in domains B and D are selected (A181V/T and N236T). Other nucleotide substitutions have been described that are instrumental for telbivudine and entecavir resistance (Fig. 78-3).

The inherent mutability of HBV indicates that single and even double polymerase mutants preexist as minor “quasispecies” even before treatment of HBV infection is begun. Because of the limitations in the sensitivities of current genotype assays, these mutants would not be detectable until they are selected and expanded under the pressure of drug treatment. Resistance to lamivudine is found in approximately 20% of patients after one year of treatment but in nearly 70% after five years (see Fig. 78-3).35 Rates of resistance to adefovir, a nucleotide analog, are 0% at one year and 29% after five years.36 The efficacy of both of these drugs against HBV is impaired by a single nucleotide substitution. The more mutations necessary for drug resistance (indicating a higher genetic barrier to resistance), the slower the emergence of and lower overall rate of resistance. For example, resistance to entecavir, another nucleoside analog, occurs in less than 1% of patients at five years because the preexistence of lamivudine-resistant mutations and one or more additional mutations in the viral polymerase gene are required for resistance.37 Persistent infection with drug-resistant HBV ultimately is associated with progression of disease and blunting of hepatic histologic improvement with antiviral therapy.38 Severe flares of hepatitis have also been reported after the emergence of drug-resistant mutants,39 and acquisition of these mutants may lead to rapidly progressive liver disease after liver transplantation (Table 78-2).40 Horizontal transmission of these mutants, which can complicate drug therapy in secondarily infected persons, is also possible.
Table 78-2 Hepatitis Flares in Patients with Chronic Hepatitis B
| CAUSE OF FLARES | COMMENT |
|---|---|
| Spontaneous | Factors that precipitate antecedent viral replication are unclear |
| Immunosuppressive therapy | Flares are often observed during withdrawal; requires preemptive antiviral therapy |
| Antiviral therapy for HBV | |
| Interferon | Flares are often observed during the second to third month; may herald virologic response |
| Lamivudine | |
| During treatment | Flares are no more common than with placebo |
| YMDD mutant | Can have severe consequences in patients with advanced liver disease |
| On withdrawal* | Flares are caused by rapid re-emergence of wild-type HBV; can have severe consequences in patients with advanced liver disease |
| HIV treatment | Flares can occur with HAART or with immune reconstitution; in addition, HBV increases the risk of antiretroviral drug hepatotoxicity |
| Genotypic variation | |
| Precore and core promoter mutants | Fluctuations in serum ALT levels are common with precore mutants |
| Superinfection with other hepatitis viruses | May be associated with suppression of HBV replication |
ALT, alanine aminotransferase; HIV, human immunodeficiency virus; HAART, highly active antiretroviral therapy; HBV, hepatitis B virus; YMDD, tyrosine-methionine-aspartate-aspartate.
PATHOGENESIS
HBV is generally not a cytopathic virus, and the severity of HBV-associated liver disease is considered to be related to the intensity of the host immunologic response to the virus. Whereas both humoral and cellular immune responses are needed for effective clearance of the virus, the cellular immune response appears to be the arm principally involved in the pathogenesis of disease. The immunologic response to HBV encompasses both an innate, or nonspecific, response (for example, natural killer cells and interferons) and an adaptive immune response, including antibodies to viral antigens, human leukocyte antigen (HLA) class II–restricted CD4+ T cells, and HLA class I–restricted CD8+ CTLs.41 Induction of the antigen-specific T-cell response is thought to occur in lymphoid organs, where the host T cells encounter viral peptide antigens (or epitopes) that are presented by antigen-presenting cells such as dendritic cells, B cells, and macrophages. This process results in the maturation and expansion of T cells that are specific for these viral epitopes and is followed by their migration to the liver, where they perform their effector function.
During acute HBV infection, most HBV DNA molecules are cleared rapidly from the liver via noncytopathic mechanisms mediated by cytokines that are released initially by cells of the innate immune system42 and later by liver-infiltrating HBV-specific CD8+ cells. Cell-mediated immune responses are efficient in self-limited infection because the responses are vigorous, multispecific, and oriented toward type 1 helper T (Th1) cells. Persons with chronic HBV infection, by contrast, exhibit infrequent, narrowly focused, and weak HBV-specific T-cell responses.43 In chronic hepatitis B, the majority of mononuclear cells in liver infiltrates of patients with chronic hepatitis B at any given time are non–antigen-specific.44
CD8+ CTLs are thought to contribute to the disease process in the liver and result in apoptosis of infected hepatocytes. To be recognized by the CD8+ CTLs, targeted hepatocytes must present viral epitopes as short peptides that have been endogenously processed and fit within the peptide-binding groove of the class I major histocompatibility complex (MHC) molecules.45 The binding of the CTL T-cell receptor (TCR) to the peptide-MHC complex on the hepatocyte surface can then result in the direct killing of the infected cell and release of potent antiviral cytokines by the activated CTL. Recognition by MHC class II–restricted CD4+ helper T cells requires the appropriate presentation of viral peptides in the context of class II MHC molecules. The CD4+ cells produce antiviral cytokines and provide help in neutralizing antibody production. Antibody neutralization limits intrahepatic spread of virus during primary infection and serves an important role in preventing reinfection.
NATURAL HISTORY
Four phases of HBV infection have been described: immune tolerance, immune clearance, the inactive carrier state, and reactivation (Fig. 78-4).
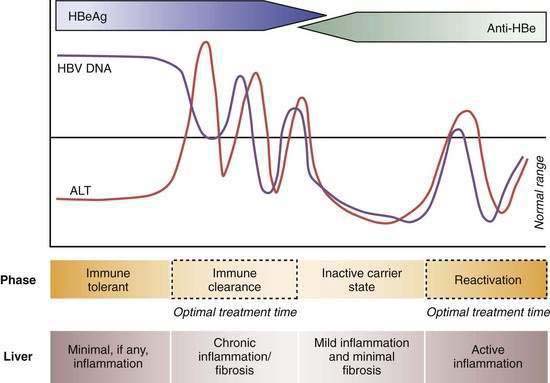
Patients who acquire the infection in the perinatal period often have high serum levels of HBV DNA without biochemical evidence of active hepatitis and are considered to be immune tolerant to HBV. When followed longitudinally, many of these patients ultimately exhibit elevated serum aminotransferase levels in association with histologic evidence of chronic hepatitis. The trigger mechanisms for this apparent change in tolerance are poorly understood but likely reflect changes in the immune reactivity of the host. Experiments in transgenic mice suggest that HBeAg induces a state of immunologic tolerance to HBV in neonates.46 Perinatal transmission of HBeAg has been considered to be a potential mechanism for the immune-tolerant state. As persons enter the immune clearance phase, HBV DNA concentrations diminish, serum alanine aminotransferase (ALT) levels rise, and hepatic histologic activity, reflecting immune-mediated lysis of infected hepatocytes, increases. The duration of this second phase varies, often lasting many years.
The third phase (inactive HBV carrier state) occurs after seroconversion from HBeAg to anti-HBe and is usually preceded by a marked reduction in serum HBV DNA to levels that are detectable only by PCR methodology, followed by normalization of serum ALT levels and resolution of liver necroinflammation. This phase may last a lifetime, but a proportion of patients ultimately undergo spontaneous or immunosuppression-mediated reactivation of HBV replication with reappearance of high levels of HBV DNA in serum, with or without HBeAg seroreversion and a rise in serum ALT levels. For unclear reasons, precore or core promoter mutants that prevent or down-regulate HBeAg production may be selected during or after HBeAg seroconversion (see earlier).47
A key event in the natural history of HBeAg-positive chronic hepatitis is seroconversion of HBeAg to anti-HBe, which is associated with marked reduction in HBV replication and biochemical and histologic remission in the majority of patients. Regression of liver fibrosis occurs gradually months to years after HBeAg seroconversion.48 Most studies have found that the mean annual rate of spontaneous HBeAg seroconversion ranges from 8% to 15% in HBV-infected children or adults with serum ALT elevations.
Longitudinal studies of untreated patients with predominantly HBeAg-positive chronic hepatitis B have shown that the frequency of development of cirrhosis ranges from 2 to 5 per 100 person-years and the five-year cumulative frequency of progression to cirrhosis from 8% to 20%.49 The rate of cirrhosis has been suggested to be higher in HBeAg-negative patients than in HBeAg-positive patients. Risk factors for the development of cirrhosis have been identified; of these, older age, the stage of fibrosis at presentation, and ongoing HBV replication with persistent or intermittent detection of HBV DNA by a non–PCR-based assay are perhaps the most important clinically. Combined infection with hepatitis D virus (HDV [see later]), HCV, or HIV and concomitant alcohol abuse have also been linked to a higher rate of development of cirrhosis.
When cirrhosis develops, two major complications may occur: hepatic decompensation and HCC. In a large European cohort with HBV-related compensated cirrhosis, the five-year cumulative frequency of hepatic decompensation was 16%, and the incidence per 100 person-years was 3.3.50 Similar rates have been reported in Asians. The cumulative five-year frequency of HCC can be as high as 14%.50 Factors associated with an increased risk of HCC include male gender, age more than 45 years, having a first-degree relative with HCC, the presence of cirrhosis, HBeAg positivity, and reversion from anti-HBe to HBeAg positivity.51 HCC can still develop in HBsAg-positive persons with none of the identified risk factors, but less frequently. In addition, HCC has been described in persons who lose HBsAg. Recommendations about ultrasonography and alpha fetoprotein screening for HCC are controversial, but in general, screening is recommended in all patients with cirrhosis and in male HBV carriers older than age 40 years in whom the likely route of transmission has been perinatal or early childhood exposure; some authorities recommend screening after age 30 years in highly viremic patients when perinatal acquisition is suspected (see Chapter 94).
ALT as a Surrogate Marker for Disease Activity
The serum ALT level has been used conventionally as a measure of disease activity in patients with chronic hepatitis B. Use of the standard reference range (0 to 40 U/L), however, can be misleading for evaluating HBV-related disease activity. A serum ALT level within the normal range is an imperfect surrogate marker for the absence of disease activity because determination of standard reference ranges has not traditionally taken into account increased body mass index (BMI), diabetes mellitus, and other features, such as alcohol intake, that tend to inflate values in a “normal” reference population (see Chapter 73). An insurance record-based study in Korea that included more than 140,000 persons who were followed for eight years demonstrated that all-cause liver-related mortality is increased when the serum ALT level exceeds 20 U/L in women and 30 U/L in men.52 This finding is particularly relevant to the management of Asians with hepatitis B and viremia. Because these patients tend to have a small body mass, a normal serum ALT value according to the standard laboratory reference range may be misleading. Such patients often have been excluded from treatment and assumed to be immune tolerant (that is, without liver disease). Studies in Asia and the United States have shown that as many as 20% to 30% of HBV carriers with persistently normal serum ALT levels and serum HBV DNA levels >104 copies/mL have stage 2 or greater (of 4) inflammation and stage 2 or greater (of 4) fibrosis on a liver biopsy specimen.53 Moreover, Asian HBV carriers with high-normal serum ALT levels (>0.5 × upper limit of normal [ULN]) have been shown to have more fibrosis on liver biopsy specimens than do those with low-normal serum ALT levels (<0.5 × ULN) and often demonstrate higher serum ALT elevations on prolonged follow-up.53–55 These data are consistent with the hypothesis that viremic HBV carriers who acquired HBV infection early in life and who have high-normal serum ALT levels may represent a subgroup of patients who have already entered the immune clearance phase and are not in the immune-tolerant phase. This occurrence should be suspected in particular in persons older than age 35 to 40 because immune tolerance often subsides after two to three decades of infection.54 Liver biopsy can be a useful tool to distinguish persons in the immune clearance phase despite normal or near-normal ALT levels but with active liver disease from those in the immune tolerant phase and absence of active liver disease (see Fig. 78-4).
HBV DNA Level and Long-Term Complications
Population-based Asian cohort studies have established that the serum HBV DNA level is the single best predictor of future progression to cirrhosis and HCC in HBV-infected persons.56,57 In a prospective cohort study, more than 3600 HBV carriers from Taiwan, of whom 60% were male, 70% were older than age 40, 85% were HBeAg negative, and 95% had normal serum ALT levels, were followed for a mean of 11 years. The calculated relative risks for cirrhosis and HCC were shown to correlate with the level of HBV DNA on entry into the study when compared with a reference population of HBV carriers in whom HBV DNA was undetectable in serum by a PCR assay.57 Even serum HBV DNA levels as low as 10,000 copies/mL (equivalent to 2000 IU/mL) were associated with a higher relative risk of cirrhosis and HCC. The relative risk of HCC was highest in persons with a serum HBV DNA level of more than 1 million copies/mL and intermediate in those in whom follow-up serum samples indicated spontaneous reduction of the serum HBV DNA level from greater than 100,000 copies/mL to less than 10,000 copies/mL. These data can be interpreted to mean that both the duration and level of viremia are important risk factors for the development of HCC. The data also suggest that suppression of serum HBV DNA levels, whether spontaneously or as a result of antiviral therapy, lowers the risk of HCC. The serum HBV DNA level remained a significant predictor of cirrhosis and HCC even after adjustment for patient age, gender, serum ALT level, and HBeAg status. Other studies in Chinese HBV carriers have demonstrated that persons in whom liver decompensation and HCC develop often have modest serum ALT elevations that are frequently below the recommended threshold (2 × ULN) for antiviral treatment in current practice guidelines (see later).58
On the basis of these data, some authorities have suggested that all persons who acquire HBV infection early in life and who are age 45 to 50 years or older and demonstrate serum HBV DNA levels of 100,000 or more copies/mL should receive long-term therapy with a nucleoside analog to prevent cirrhosis and HCC.59,60 In a landmark study,61 more than 600 Asian patients with advanced fibrosis and a serum HBV DNA level greater than 100,000 copies/mL were randomized in a ratio of 2 : 1 to active treatment with lamivudine or placebo. Treatment was planned for 5 years, but the study was discontinued after a mean duration of only 32 months because disease progression and HCC occurred significantly more frequently in the group of patients randomized to placebo.61 Therapeutic benefit was independent of the serum ALT level at baseline. These data strongly suggest that continuing suppression of serum HBV DNA by antiviral therapy can alter the long-term outcome of active chronic hepatitis B.
CLINICAL FEATURES
Acute Hepatitis B
The incubation period of acute hepatitis B varies from a few weeks to 6 months (average, 60 to 90 days), depending on the amount of replicating virus in the inoculum. The disease may be more severe in patients coinfected with other hepatitis viruses and in those with established underlying liver disease.62 Abstention from alcohol is usually recommended, but the chance of an uneventful recovery does not appear to be affected by the consumption of moderate amounts of alcohol (20 to 30 g daily) during the convalescent phase.63 Acute infections are heralded by a serum sickness–like prodrome of fever, arthralgia or arthritis, and rash, which is most commonly maculopapular or urticarial, in 10% to 20% of patients. This prodrome results from circulating HBsAg–anti-HBs complexes that activate complement and are deposited in the synovium and walls of cutaneous blood vessels.64 These features generally abate before the manifestations of liver disease and peak serum aminotransferase elevations are observed. Jaundice develops in only about 30% of patients.
Clinical symptoms and jaundice generally disappear after one to three months, but some patients have prolonged fatigue even after serum ALT levels return to normal. In general, elevated serum ALT levels and serum HBsAg titers decline and disappear together, and in approximately 80% of cases, HBsAg disappears by 12 weeks after the onset of illness.65 In 5% to 10% of cases, HBsAg is cleared early and is no longer detectable by the time the patient first presents to a health care provider. Persistence of HBsAg after six months implies development of a carrier state, with only a small likelihood of recovery during the next 6 to 12 months. Delayed clearance of HBsAg has been reported to be preceded by a decline in HBsAg titers.
After clinical recovery from acute hepatitis B and HBsAg seroconversion, HBV DNA often remains detectable in serum as determined by a PCR assay (see later discussion of diagnosis). After resolution of acute hepatitis, the numbers of HBV-specific CD4+ and CD8+ cells in blood and liver decrease rapidly. Nonetheless, T-cell responsiveness remains high on re-encounter with HBV antigens, indicating that traces of virus can maintain the CTL response indefinitely after clinical recovery, thereby exerting control over the virus and preventing reactivated infection.41,66
Fulminant hepatitis occurs in less than 1% of cases (see Chapter 93). Fulminant hepatitis B generally occurs within four weeks of the onset of symptoms and is associated with encephalopathy, multiorgan failure, and a high mortality rate (>80%) if not treated by liver transplantation. Patients older than age 40 years appear to be more susceptible than younger persons to “late-onset liver failure,” in which encephalopathy, renal dysfunction, and other extrahepatic complications of severe liver insufficiency become manifest over the course of several months. The pathogenic mechanisms of fulminant hepatitis are poorly understood but are presumed to involve massive immune-mediated lysis of infected hepatocytes. This proposed mechanism may explain why many patients with fulminant hepatitis B have no evidence of HBV replication in serum at presentation.
Chronic Hepatitis B
Physical findings may be normal, or hepatosplenomegaly may be found. In decompensated cirrhosis, spider angiomata, jaundice, ascites, and peripheral edema are common. Liver biochemical test results are usually completely normal during the inactive HBV carrier state. In contrast with patients in the immune-tolerant phase of HBV infection, most patients in the immune-clearance phase of chronic HBV infection have mild to moderate elevations in serum AST and ALT levels. During exacerbations of disease, serum ALT levels may be as high as 1000 U/L or more, and the clinical and laboratory picture is indistinguishable from that of acute hepatitis B, including the presence in serum of IgM anti-HBc. Progression to cirrhosis should be suspected whenever hypersplenism, hypoalbuminemia (in the absence of nephropathy), or prolongation of the prothrombin time is found. The serum AST level is typically higher than the serum ALT level in patients with advanced cirrhosis (see Chapter 73).
Extrahepatic Manifestations
Extrahepatic syndromes seen in association with acute or chronic hepatitis B are important to recognize because they may occur without clinically apparent liver disease and can be mistaken for independent disease processes in other organ systems. The pathogenesis of these extrahepatic disorders has not been fully elucidated but likely involves an aberrant immunologic response to extrahepatic viral proteins.67 Many of the extrahepatic manifestations (e.g., arthritis, dermatitis, glomerulonephritis, polyarteritis nodosa, cryoglobulinemia, papular acrodermatitis, and polymyalgia rheumatica) are observed in association with circulating immune complexes that activate serum complement. Antiviral therapy may be indicated for persistent symptoms.
Glomerulonephritis
Several types of glomerular lesions have been described in patients with chronic HBV infection; membranous glomerulonephritis and membranoproliferative glomerulonephritis are the most common.68 Renal biopsy specimens have demonstrated immune complex deposition and cytoplasmic inclusions in the glomerular basement membrane. The immune complexes activate complement and production of cytokines with a subsequent inflammatory response. Nephrotic syndrome is the most common presentation of HBV-associated glomerulonephritis. In affected children, renal failure at presentation is almost always mild, and a history of clinical liver disease is uncommon. Nevertheless, liver biopsy specimens almost always demonstrate varying degrees of chronic hepatitis. The diagnosis of HBV-associated glomerulonephropathy is usually established by serologic evidence of HBV antigens or antibodies, the presence of immune-complex glomerulonephritis in a renal biopsy specimen, and the demonstration of glomerular deposits of one or more HBV antigens, such as HBsAg, HBcAg, or HBeAg, by immunohistochemistry. Most patients have detectable HBeAg in serum and, in addition, demonstrate low serum C3 and occasionally low C4 levels. The renal disease typically resolves in months to several years, especially in children. Often, resolution occurs in conjunction with HBeAg seroconversion. Rarely, however, renal failure may ensue. The natural history of HBV-related glomerulonephritis in adults has not been well defined, but several reports suggest that glomerular disease is often slowly and relentlessly progressive.69 Successful treatment has been accomplished with interferon alpha and has been linked to long-term control of HBV replication.70 Therapy with nucleoside analogs has resulted in improved renal function and diminished proteinuria.
Cryoglobulinemia
Type II cryoglobulins consist of a polyclonal IgG and monoclonal IgM, whereas type III cryoglobulins contain polyclonal IgG and rheumatoid factor. Type II and type III cryoglobulinemia have been associated with hepatitis B, but the association is uncommon. In a large patient cohort, the frequency of cryoglobulinemia was significantly higher in patients with chronic HCV infection (54%) than in patients with chronic HBV infection (15%) (see Chapter 79). Cryoglobulinemia may be associated with systemic vasculitis (purpura, arthralgias, peripheral neuropathy, and glomerulonephritis) but is often paucisymptomatic or asymptomatic. Interferon has been used successfully to treat symptomatic cryoglobulinemia in association with chronic hepatitis B. Experience with nucleoside analog therapy has not been reported.
Histopathologic Features
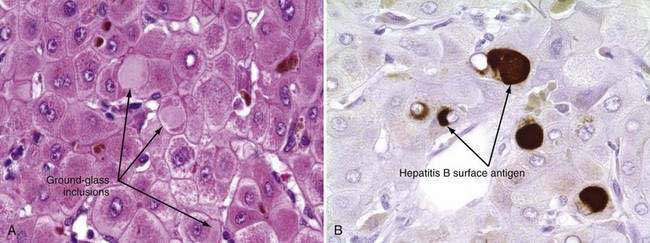
The only histologic feature noted on routine light microscopy that is specific for chronic hepatitis B is the presence of ground-glass hepatocytes (Fig. 78-5). This morphologic finding results from accumulation of HBsAg particles (20 to 30 nm in diameter) in the dilated endoplasmic reticulum. Because of high levels of cysteine in HBsAg, ground-glass cells have a high affinity for certain dyes, such as orcein, Victoria blue, and aldehyde fuchsin. Ground-glass hepatocytes also may be seen in HBV carriers, in whom they may be detected in up to 5% of cells. When present in abundance, ground-glass hepatocytes often indicate active viral replication.71 Immunofluorescence and electron microscopic studies have shown HBcAg inside the hepatocyte nuclei of affected cells. During periods of intense hepatitis activity, cytoplasmic core antigen staining is generally observed. After successful treatment of HBV infection with a nucleoside analog, the cytoplasmic core antigen staining often disappears, but nuclear core antigen staining may remain, indicating persistence of the HBV cccDNA template.
Acute Flares in Chronic Hepatitis B
Chronic hepatitis B is often punctuated by sudden flares of disease activity that are reflected by a rise in serum aminotransferase levels. Although a uniform definition is lacking, a flare has frequently been described as a rise in serum ALT levels to at least two times the baseline value. Spontaneous flares are an important part of the natural history of hepatitis B because when they occur repeatedly, they lead to histologic progression. Acute flares in chronic hepatitis B occur in association with a number of circumstances and clinical situations (see Table 78-2). Most flares result from a change in the balance between immunologic responses to HBV and the level of viral proliferation. Acute flares in chronic hepatitis B that are not explainable by infection with other hepatotropic viruses often occur as a secondary response to increased levels of replicating wild-type or mutant HBV or as a result of therapeutic intervention with immunologic modifiers such as interferon, glucocorticoids, and cancer chemotherapy. In some instances, the event that initiates an acute exacerbation of chronic hepatitis B may not be readily identifiable, and the flare is considered spontaneous.
Spontaneous Flares
Spontaneous exacerbations of chronic hepatitis B often result from reactivated infection, and an increase in serum HBV DNA levels often precedes an increase in serum aminotransferase levels. Histologic evidence of acute lobular hepatitis superimposed on the changes of chronic viral hepatitis is frequently observed during these flares.72 IgM anti-HBc, a marker that is often diagnostic of acute viral hepatitis, may also appear in serum at this time.73
The reasons for reactivated infection are unknown but likely relate to subtle changes in the immunologic control of viral replication. Reactivation seems to occur more commonly in persons who are infected with HIV.74 In persons who acquire HBV infection early in life, flares become more common during adulthood, presumably because of a breakdown in immune tolerance to HBV.75
Flares also can occur in patients who are in the replicative phase of infection (i.e., already positive for HBV DNA and HBeAg in serum). In these instances, HBV replication intensifies, serum HBV DNA levels rise, and liver biochemical deterioration occurs, often without the subsequent loss of HBeAg. Multiple episodes of reactivation and remission have been shown to accelerate the progression of chronic hepatitis B and are particularly likely to occur in patients infected with the precore mutant form of chronic hepatitis B (see earlier).47
Immunosuppressive Therapy-Induced Flares
Reactivation of HBV replication is a well-recognized complication in patients with chronic HBV infection who receive cytotoxic or immunosuppressive therapy.76 Suppression of the normal immunologic responses to HBV leads to enhanced viral replication and is thought to result in widespread infection of hepatocytes by HBV. On discontinuation of immunosuppressive medications, such as cancer chemotherapy, antirejection drugs, and glucocorticoid therapy, immune competence is restored and infected hepatocytes are rapidly destroyed. The more potent the immunosuppression, the higher the level of viral replication and, thus, the greater the potential for serious clinical consequences of sudden withdrawal of the therapy and restoration of immunologic competence. Postmortem studies of liver tissue from patients with severe liver injury have documented sparse staining of viral antigens, suggesting that the patients were in an active state of immune clearance.77
The vast majority of patients who experience immunosuppressive therapy–induced flares have been positive for HBsAg in serum before treatment, but some studies have described the reappearance of HBsAg in patients who were initially positive for anti-HBs, anti-HBc, or both.78 Reactivated hepatitis in patients who are negative for HBsAg and positive for either anti-HBc or anti-HBs is explainable by the possible latency of HBV in liver and mononuclear cells and the large extrahepatic reservoir of HBV. Chemotherapy given to patients with cancer who are HBV carriers is associated with an increased risk of liver-related morbidity and mortality.79
Reactivated hepatitis B also occurs in patients who are given immunosuppressive medications to prevent organ transplant rejection. The frequency of reactivated hepatitis appears to be particularly high in patients who undergo bone marrow transplantation because of extensive immunologic conditioning before transplantation and treatment of graft-versus-host disease.80 Rarely, fibrosing cholestatic hepatitis, a rapidly progressive form of liver injury associated with inordinately high levels of HBsAg and HBcAg in liver tissue, may develop in such patients.81
Acute flares of hepatitis B resulting from cancer chemotherapy and other immunosuppressive drugs are often detected after substantial increases in serum aminotransferase levels have been noted. Initiation of antiviral treatment after detection of such biochemical abnormalities has little effect on reducing liver injury because much of the immunologic response to HBV and viral elimination has already occurred. Instead, the key to management lies in anticipating the occurrence of a flare, initiating antiviral treatment preemptively (e.g., 4 to 6 weeks before the start of chemotherapy), and continuing the treatment for 6 to 12 months after completion of chemotherapy.82
Antiviral Therapy–Induced Flares
Interferon
Interferon-induced flares of chronic hepatitis B occur in approximately one third of treated patients and result from the immunostimulatory properties of the drug. Flares generally occur during the second or third month of treatment with conventional preparations of interferon. Flares also occur in patients treated with pegylated interferon and have been reported to occur more frequently in patients infected with HBV genotype A than with other genotypes. This finding may explain the higher rate of sustained virologic response and HBsAg clearance in patients infected with HBV genotype A.83 Serum ALT flares have been shown to be a predictor of sustained virologic response, particularly in patients with high levels of viremia.84 Flares tend to be particularly common in patients who have decompensated liver disease, with rates as high as 50% reported in one series.85 Such flares are frequently associated with clinical deterioration in the patient.
Nucleoside Analogs
Serum ALT flares occur in approximately 20% to 25% of patients after withdrawal of nucleoside analogs such as lamivudine and adefovir. These flares probably are caused by rapid resurgence of wild-type HBV, and although generally well tolerated, they have been associated with serious clinical exacerbations in patients with advanced liver disease.86 Reinstitution of therapy is often associated with a decline in serum HBV DNA and aminotransferase levels. Flares have been seen to follow the emergence of YMDD mutant HBV during therapy with lamivudine (see earlier).87
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


