CHAPTER 92 Hepatic Encephalopathy, Hepatorenal Syndrome, Hepatopulmonary Syndrome, and Systemic Complications of Liver Disease
HEPATIC ENCEPHALOPATHY
The term hepatic encephalopathy (HE) encompasses a wide array of transient and reversible neurologic and psychiatric manifestations usually found in patients with chronic liver disease and portal hypertension, but also seen in patients with acute liver failure. HE develops in 50% to 70% of patients with cirrhosis, and its occurrence is a poor prognostic indicator, with projected one- and three-year survival rates of 42% and 23%, respectively, without liver transplantation.1 Symptoms may range from mild neurologic disturbances to overt coma.2,3 HE is often triggered by an inciting event that results in a rise in the serum ammonia level. The precise underlying pathophysiologic mechanisms are not well understood, and the mainstay of therapy is the elimination of the precipitating event and excess ammonia.4 Liver transplantation generally reverses HE.
PATHOPHYSIOLOGY
A number of factors, occurring alone or in combination, have been implicated in the development of HE. These factors may differ in acute and chronic liver disease and include the production of neurotoxins, altered permeability of the blood-brain barrier, and abnormal neurotransmission (Fig. 92-1). The best-described neurotoxin involved in HE is ammonia, which is produced primarily in the colon, where bacteria metabolize proteins and other nitrogen-based products into ammonia. Enterocytes synthesize ammonia from glutamine.4–6 Once produced, ammonia enters the portal circulation and, under normal conditions, is metabolized and cleared by hepatocytes. In cirrhosis and portal hypertension, reduced hepatocyte function and portosystemic shunting contribute to increased circulating ammonia levels. Arterial hyperammonemia is observed in up to 90% of patients with HE, although serum levels are neither sensitive nor specific indicators of its presence. Increased permeability of the blood-brain barrier increases the uptake and extraction of ammonia by the cerebellum and basal ganglia.7–9 Acute hyperammonemia appears to have a direct effect on brain edema, astrocyte swelling, and the transport of neurally active compounds such as myoinositol, and thereby contributes to HE.10–12
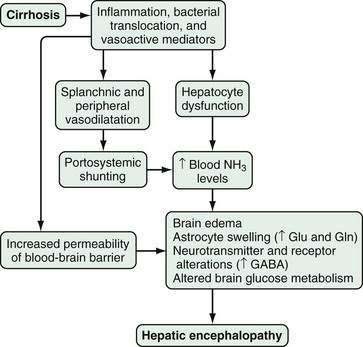
Figure 92-1. Pathophysiology of hepatic encephalopathy. GABA, γ-aminobutyric acid; Gln, glutamine; Glu, glutamate; NH3, ammonia.
Other alterations in HE affect neuronal membrane fluidity, central nervous system (CNS) neurotransmitter expression, and neurotransmitter receptor expression and activation.13,14 The γ-aminobutyric acid (GABA)–benzodiazepine system has been the most well studied. Although CNS benzodiazepine levels and GABA receptor concentrations are unchanged in animal models of HE, increased sensitivity of the astrocyte (peripheral-type) benzodiazepine receptor enhances activation of the GABA-benzodiazepine system.15,16 This activation occurs in part through a feed-forward system in which production of neurosteroids (allopregnanolone and tetrahydrodeoxycorticosterone) by astrocytes further activates the GABAA-benzodiazepine receptor system.17,18 Other factors that influence CNS neurotransmission, including serotonin (5-hydroxytryptamine, 5-HT),19–21 nitric oxide (NO), circulating opioid peptides, manganese, and increased oxygen free radical production, have also been postulated to contribute to HE.4
Finally, hyperammonemia, particularly in acute liver failure, also increases astrocyte glutamine production via glutamine synthetase. The rise in astrocyte glutamine and glutamate concentrations contributes to factors associated with CNS dysfunction.5,22,23
CLINICAL FEATURES AND DIAGNOSIS
HE may present as a spectrum of reversible neuropsychiatric symptoms and signs, ranging from mild changes in cognition to profound coma, in patients with acute or chronic liver disease. It is often precipitated by an inciting event (e.g., gastrointestinal bleeding, electrolyte abnormalities, infections, medications, dehydration). The diagnosis of HE, therefore, requires careful consideration in the appropriate clinical situation. Occasionally, HE may be the initial presentation of chronic liver disease. Subtle findings in HE may include forgetfulness, alterations in handwriting, difficulty with driving, and reversal of the sleep-wake cycle.24,25 Overt findings may include asterixis, agitation, disinhibited behavior, seizures, and coma. Other causes of altered mental status, particularly hypoglycemia, hyponatremia, medication ingestion, and structural intracranial abnormalities resulting from coagulopathy or trauma, should be considered and rapidly excluded in patients suspected of having HE.
No specific laboratory findings indicate the presence of HE definitively. The most commonly used test to assess a patient with possible HE is the blood ammonia level. An elevation in the blood ammonia level in a patient with cirrhosis and altered mental status supports a diagnosis of HE. Blood ammonia levels may be elevated in the absence of HE, however, because of gastrointestinal bleeding or the ingestion of certain medications (e.g., diuretics, alcohol, narcotics, valproic acid).15,26,27 In addition, blood ammonia levels may be elevated in the presence of HE, even in the absence of cirrhosis and portal hypertension, in patients with metabolic disorders that influence ammonia generation or metabolism, such as urea cycle disorders (see Chapter 76) and disorders of proline metabolism (Table 92-1).28,29 Use of a tourniquet when blood is drawn and delayed processing and cooling of a blood sample may raise the blood ammonia level.15 Measurement of arterial ammonia offers no advantage over measurement of venous ammonia levels in patients with chronic liver disease.30 In patients with acute liver failure, however, elevated arterial ammonia levels (150 to 200 mg/dL or higher) may be predictive of the presence of brain edema and herniation (see Chapter 93).12,31,32
Table 92-1 Differential Diagnosis of Hyperammonemia
Of the scoring systems used to grade the severity of HE, the West Haven system, based on a scale of 0 to 4, is the most widely used in clinical practice (Table 92-2).25 Although clinically useful, the West Haven criteria are insensitive and have led to the development of standardized psychometric tests and rapid bedside mental status assessments to aid in the diagnosis of HE and facilitate research.33–37 One simple paper and pencil test, the portosystemic encephalopathy syndrome test (PSET), evaluates the patient’s attention, concentration, fine motor skills, and orientation and has been shown to be highly specific for the diagnosis of HE.33,38 The development of these tests has led to recognition of the syndrome of minimal HE, in which abnormalities are observed on testing but clinically recognizable alterations of HE are minimal or not detected. The presence of minimal HE is common in patients with cirrhosis, appears to influence the patient’s quality of life and driving ability, and confers an increased risk that overt HE will develop in the patient. Whether treatment of minimal HE confers any benefit is an area of active investigation.24,39–41
Table 92-2 Clinical Stages of Hepatic Encephalopathy
| GRADE | Impairment | |
|---|---|---|
| INTELLECTUAL FUNCTION | NEUROMUSCULAR FUNCTION | |
| 0 | Normal | Normal |
| Minimal, subclinical | Normal examination findings. Subtle changes in work or driving | Minor abnormalities of visual perception or on psychometric or number tests |
| 1 | Personality changes, attention deficits, irritability, depressed state | Tremor and incoordination |
| 2 | Changes in sleep-wake cycle, lethargy, mood and behavioral changes, cognitive dysfunction | Asterixis, ataxic gait, speech abnormalities (slow and slurred) |
| 3 | Altered level of consciousness (somnolence), confusion, disorientation, and amnesia | Muscular rigidity, nystagmus, clonus, Babinski sign, hyporeflexia |
| 4 | Stupor and coma | Oculocephalic reflex, unresponsiveness to noxious stimuli |
Modified from the West Haven Criteria; in Ferenci P, Lockwood A, Mullen K, et al. Hepatic encephalopathy—definition, nomenclature, diagnosis, and quantification: Final report of the working party at the 11th World Congresses of Gastroenterology, Vienna, 1998. Hepatology 2002; 35:716-21.
A number of novel imaging and functional tests have been studied in the diagnosis of HE. Magnetic resonance spectroscopy (MRS) has been used to measure brain concentrations of choline and glutamine noninvasively.42 Magnetic resonance (MR) T1 mapping with partial inversion recovery (TAPIR) has been investigated as a means to measure changes in the brain quantitatively over clinically relevant measurement times.33 Whether MR-based techniques can be standardized and become practical diagnostic tests is uncertain. The critical flicker frequency test, a simple light-based test that has been used to assess cerebral cortex function in a number of disorders, has been shown to be a reliable marker of minimal HE and may become a clinically useful screening test.34–36
TREATMENT
Current treatments for HE are directed primarily toward the elimination or correction of precipitating factors (e.g., bleeding, infection, hypokalemia, medications, dehydration), reduction in elevated blood ammonia levels, and avoidance of the toxic effects of ammonia in the CNS. In the past, dietary protein restriction was considered an important component of the treatment of HE. Subsequent work, however, has suggested that limiting protein-calorie intake is not beneficial in patients with HE.43–45 Vegetable and dairy proteins are preferred to animal proteins because of a more favorable calorie-to-nitrogen ratio. Although branched-chain amino acid supplementation may improve symptoms modestly, the benefits of such supplementation are not sufficient to justify its routine use.4
Nonabsorbable disaccharides have been the cornerstone of the treatment of HE. Oral lactulose or lactitol (the latter is not available in the United States) are metabolized by colonic bacteria to byproducts that appear to have beneficial effects by causing catharsis and reducing intestinal pH, thereby inhibiting ammonia absorption.46 These agents improve symptoms in patients with acute and chronic HE when compared with placebo but do not improve psychometric test performance or mortality. The most common side effects experienced by patients who take lactulose are abdominal cramping, flatulence, diarrhea, and electrolyte imbalance. Lactulose may also be administered per rectum (as an enema) to patients who are at increased risk of aspiration, although the efficacy of enema administration has not been evaluated.
Oral antibiotics also have been used to treat HE, with the aim of modifying the intestinal flora and lowering stool pH to enhance the excretion of ammonia. Antibiotics are generally used as second-line agents after lactulose or in patients who are intolerant of nonabsorbable disaccharides. Neomycin has been approved by the U.S. Food and Drug Administration (FDA) for use in acute HE in a dose of 1 to 3 g orally every six hours for up to six days but has been used more commonly off-label to treat chronic HE in doses of 0.5 to 1 g every 12 hours, in addition to lactulose. The efficacy of neomycin in acute or chronic HE, however, is not clearly established,47 and ototoxicity and nephrotoxicity caused by neomycin have been reported, particularly in patients with preexisting renal dysfunction.4 Rifaximin has been studied and approved by the FDA for the treatment of chronic HE on the basis of the results of a multicentered, randomized, controlled trial in which the overall clinical efficacy and rate of side effects were similar in patients treated with lactitol and those treated with rifaximin.48 The usual dose is 400 mg orally three times daily. Two systematic reviews of randomized controlled trials that compared rifaximin with other therapies (nonabsorbable disaccharides and other antibiotics) for the treatment of acute or chronic HE have confirmed that the efficacy and side effect profiles are comparable.49,50 Other antibiotics, including metronidazole and vancomycin, have been reported to be effective in small trials and case series, but the data to support their use are insufficient.
In addition to antibiotics, several other agents that may modify intestinal flora and modulate ammonia generation or absorption have been evaluated as potential treatments for HE. Acarbose, an intestinal α-glucosidase inhibitor used to treat type 2 diabetes mellitus, inhibits the intestinal absorption of carbohydrates and glucose and results in their enhanced delivery to the colon. As a result, the ratio of saccharolytic to proteolytic bacterial flora is increased, and blood ammonia levels are decreased. A randomized, controlled, double-blind, crossover trial has demonstrated that acarbose improves mild HE in patients with cirrhosis and adult-onset diabetes mellitus.51 Similarly, probiotic regimens have been used to modify intestinal flora and diminish ammonia generation. Four small studies have suggested that these agents may be beneficial in humans with mild HE.40,52–55 These agents merit further evaluation and may be alternatives for patients who do not tolerate lactulose.
Strategies to enhance ammonia clearance may also be useful in the treatment of HE. Sodium benzoate, sodium phenylbutyrate, and sodium phenylacetate, all of which increase ammonia excretion in urine, are approved by the FDA for the treatment of hyperammonemia resulting from urea cycle enzyme defects and may improve HE in cirrhosis (see Chapter 76). Administration of sodium benzoate, however, results in a high sodium load, and the efficacy of this agent is not clearly established.4,56 The combination of intravenous sodium phenylacetate and sodium benzoate (Ammonul, Ucyclyd Pharma, Scottsdale, Ariz) in HE is being studied. Administration of zinc, which has been used because zinc deficiency is common in patients with cirrhosis57–59 and because zinc increases the activity of ornithine transcarbamylase, an enzyme in the urea cycle, may also improve HE; however, clear efficacy has not been established. Extracorporeal albumin dialysis using the molecular adsorbent recirculating system (MARS) has resulted in a reduction in blood ammonia levels and improvement in severe HE in patients with acute-on-chronic liver failure (see Chapter 93).60 Further studies are needed to clarify whether albumin dialysis has a role in treatment of HE. Finally, l-ornithine–l-aspartate (LOLA), a salt of the amino acids ornithine and aspartic acid that activates the urea cycle and enhances ammonia clearance, has been shown in several randomized controlled studies to improve HE compared with lactulose61–63; however, this agent is not available in the United States.
Flumazenil is a specific benzodiazepine (GABAA receptor) antagonist that has been used in patients with HE. It improves the degree of encephalopathy and electrophysiologic findings in approximately one fourth of patients with grade 3 or 4 HE. It has a short half-life and a number of potential side effects, including seizures, arrhythmias, and withdrawal symptoms, that limit its clinical usefulness.4
HEPATORENAL SYNDROME
The term hepatorenal syndrome (HRS) was first used in 1932 to describe acute kidney injury, mainly acute tubular necrosis (ATN) or interstitial nephritis, in a group of patients who had undergone biliary tract surgery.64 As pathophysiologic mechanisms were better elucidated, HRS was found to be part of a cascade of events associated with intense dilatation of the splanchnic arterial vasculature in the setting of cirrhosis or acute liver injury and resulting in profound renal arterial vasoconstriction and progressive renal failure.65 Histologically, the kidneys are normal in HRS. Function may be restored by correction of portal hypertension, liver transplantation, removal of the kidneys and transplantation of them into a noncirrhotic recipient, and, in some cases, medical therapy (see later).66–68
Acute renal dysfunction occurs in 15% to 25% of hospitalized patients with cirrhosis. Among the multiple causes of acute kidney injury, prerenal azotemia (resulting from intravascular volume depletion) is most common, accounting for 60% to 80% of cases. ATN is the second most common cause of acute kidney injury in this setting and accounts for 20% to 40% of cases.69,70 HRS appears to be an extension of the pathophysiology of prerenal azotemia and is therefore potentially reversible. The annual frequency of HRS in cirrhotic patients with ascites is roughly 8%71 and, in some reports, as high as 40%.72 HRS develops in approximately 30% of cirrhotic patients who are admitted with spontaneous bacterial peritonitis (SBP) or other infection, 25% who are hospitalized with severe alcoholic hepatitis, and 10% who require serial large-volume paracentesis.73 The observation that morbidity and mortality remain high once the syndrome is established has led to a focus on the prevention and early therapy of renal dysfunction in patients with cirrhosis.71
PATHOPHYSIOLOGY
The pathophysiology of HRS is complex and incompletely characterized. Three important components are recognized to contribute to the initiation and perpetuation of altered renal perfusion (Fig. 92-2): (1) arterial vasodilatation in the splanchnic and systemic circulation; (2) renal vasoconstriction; and (3) cardiac dysfunction.74 These components influence renal function in concert and form the basis for current therapies and preventive strategies.
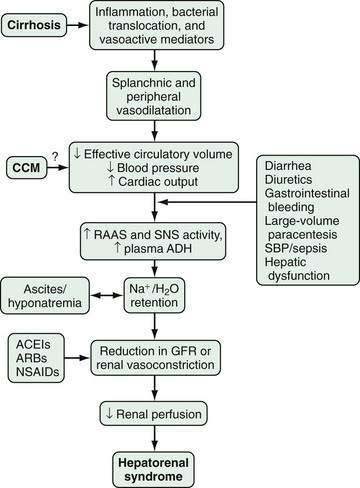
Splanchnic Arterial Vasodilatation
Splanchnic and systemic arterial vasodilation are hallmarks of the progression of portal hypertension in cirrhosis and lead to decreased effective circulating blood volume and ultimately to a decrease in blood pressure. This process is mediated by a number of endogenous substances, including NO, carbon monoxide (CO), glucagon, prostacyclin, adrenomedullin, and endogenous opiates that are released or act locally in the vasculature in response to mechanical and inflammatory signals.65,71,75,76 In the early stages of portal hypertension, compensation for the decrease in effective circulatory volume includes an increase in heart rate and cardiac output, thereby creating a hyperdynamic circulation.77 As liver disease and splanchnic vasodilatation progress, additional compensatory mechanisms are activated.
Renal Arterial Vasoconstriction
Splanchnic and systemic vasodilatation also lead to compensatory renal vasoconstriction and renal sodium and water retention. These responses are mediated by stimulation of the sympathetic nervous system, activation of the renin-angiotensin-aldosterone system (RAAS), and nonosmotic release and activity of arginine vasopressin (as a result of increased secretion and decreased clearance of arginine vasopressin and apparent increased expression of vasopressin-regulated water channels), as well as intrarenal events. Although the precise intrarenal mechanisms are speculative, altered production or action of endothelins, prostaglandins, kallikreins, and F2-isoprostanes may contribute to renal vasoconstriction.72,78,79 Ultimately, the balance between vasoconstrictive responses in the kidney and systemic and splanchnic vasodilatation is lost, thus leading to a prominent increase in renal vascular resistance, decrease in renal perfusion, and reduction in the glomerular filtration rate.71,77
Cardiac Dysfunction
Impaired cardiac function also may contribute to renal hypoperfusion in HRS. In one prospective study, HRS developed in cirrhotic patients with more severe arterial vasodilatation and lower cardiac output.80 In another study of a cohort of patients who were treated for SBP, renal dysfunction (including HRS in some cases) developed in those with lower cardiac output and lower arterial pressure measurements associated with higher circulating levels of norepinephrine and renin plasma activity, despite effective treatment of the infection.81 These data demonstrate that cardiac output is impaired in patients with cirrhosis in whom HRS develops as compared with those in whom HRS does not develop, and suggests that cardiac dysfunction may be an important additional factor in the pathogenesis of HRS. The relationship between cardiac dysfunction in patients with HRS and cirrhotic cardiomyopathy (see later) has not been studied.82,83
CLINICAL FEATURES AND DIAGNOSIS
HRS is a functional disorder, and therefore diagnostic laboratory and imaging studies are not available. The diagnosis of HRS requires a high index of clinical suspicion and exclusion of other potential causes of kidney injury. The diagnostic criteria for HRS as defined by the International Ascites Club Consensus Workshop in 2007 include the following: (1) cirrhosis with ascites; (2) serum creatinine level higher than 1.5 mg/dL (133 µmol/L); (3) lack of improvement in the serum creatinine level to 1.5 mg/dL (133 µmol/L) or less after at least two days of diuretic withdrawal and volume expansion with albumin (1 g/kg of body weight/day, up to a maximum of 100 g/day); (4) absence of shock, (5) lack of current or recent treatment with nephrotoxic drugs; and (6) absence of parenchymal kidney disease as indicated by proteinuria of more than 500 mg/day, microhematuria (>50 red blood cells/high power field), or abnormal renal ultrasonographic findings (Table 92-3).74
Table 92-3 Diagnostic Criteria for Hepatorenal Syndrome*
* As defined by the International Ascites Club Consensus Workshop in 2007 (Salerno F, Gerbes A, Ginès P, et al. Diagnosis, prevention and treatment of hepatorenal syndrome in cirrhosis. Gut 2007; 56:1310-8).
Several aspects of the diagnosis of HRS deserve emphasis. First, in patients with no prior evidence or history of renal impairment, the diagnostic criteria for HRS include an increase in the serum creatinine level by 50% above baseline to a level higher than 1.5 mg/dL (133 µmol/L).71 Although this definition is standardized, a subset of patients with cirrhosis and end-stage liver disease have a profound decrease in muscle mass and urea synthesis that may in turn result in reduced serum creatinine and blood urea nitrogen levels, thereby potentially delaying recognition of HRS.73,84 Second, many medications, most notably diuretics, lactulose, angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, and nonsteroidal anti-inflammatory drugs, may influence intravascular volume status and renal perfusion and should be identified expeditiously and discontinued in the setting of acute renal dysfunction. Third, even though SBP may not be accompanied by obvious symptoms and signs, HRS may develop in as many as 20% of affected patients.85 Therefore, a low threshold for evaluating cirrhotic patients with ascites for the presence of SBP is required.
CLASSIFICATION
HRS has traditionally been classified into two types on the basis of clinical characteristics and prognosis (types 1 and 2).74 Some authors have advocated expanding the classification to include patients who are not encompassed within the current framework (i.e., type 3 HRS).72
Type 1 HRS presents as a rapidly progressive form of renal dysfunction. Typically, the serum creatinine level doubles to a value higher than 2.5 mg/dL in a period of two weeks or less. Type 1 HRS is often triggered by an inciting event that causes a rapid decline in the hemodynamic parameters that maintain renal homeostasis in cirrhotic patients.70 The most common triggers include severe bacterial infections,86–88 gastrointestinal bleeding, surgical procedures, and acute liver injury.74,89,90 SBP is the main bacterial infection that predisposes cirrhotic patients to develop HRS. Other bacterial infections have less of an impact on the development of the syndrome, unless sepsis is present or the response to antibiotic therapy is poor.71,87,91 Patients who exhibit high levels of inflammatory response markers and patients with severe circulatory depression prior to the onset of infection are most susceptible to the development of HRS. Some degree of adrenal insufficiency has been found in 80% of patients with septic shock in whom HRS develops, and administration of glucocorticoids may improve survival.92
Type 2 HRS is a more slowly progressing entity compared with type 1 HRS but still carries a median survival of only approximately six months. Type 2 HRS is observed in patients with severe ascites (diuretic resistant) and is characterized by serum creatinine levels lower than 2.5 mg/dL. The degree of arterial hypotension and circulatory dysfunction is less than that seen with type 1 HRS. Type 1 HRS may develop in patients with type 2 HRS following a triggering event.71,72
Many patients with cirrhosis and portal hypertension also have underlying chronic kidney disease, which complicates recognition of HRS, even in the presence of underlying pathophysiologic mechanisms that favor the development of HRS. Whether these patients should be considered to have a unique form of HRS (type 3) and whether they should be treated in a fashion similar to that for other patients with HRS have not been clearly defined.72
PREVENTION AND TREATMENT
The high mortality rate of HRS underscores the importance of prevention. In particular, intravascular volume depletion (resulting from overdiuresis, diarrhea caused by lactulose, gastrointestinal bleeding from gastroesophageal varices, or large-volume paracentesis without colloid administration), nephrotoxins (e.g., nonsteroidal anti-inflammatory drugs, nephrotoxic antibiotics), and infection (SBP, bacteremia) should be avoided or addressed if present. Specific guidelines address the primary and secondary prophylaxis of variceal bleeding (see Chapter 90), administration of colloid (albumin) to patients with a rising serum creatinine level after a large-volume paracentesis and in the presence of SBP (see Chapter 91), and prophylactic administration of antibiotics to patients at high risk of SBP or other infections because of hospitalization for gastrointestinal bleeding (see Chapters 19 and 90).85,86,89,91 Routine invasive hemodynamic monitoring of cirrhotic patients with a rising serum creatinine level does not have a clear benefit and is not recommended.
The concept that treatment of HRS is possible and may improve survival has emerged since 2000. Current options include medical therapies, transjugular intrahepatic portosystemic shunt (TIPS) placement, and liver transplantation. Medical therapies for HRS are directed toward reversing the underlying splanchnic and systemic vasodilatation with vasoconstrictors and increasing effective circulatory volume with the use of colloid. Such treatment is used increasingly as a temporizing measure until definitive treatment for liver disease (liver transplantation) or portal hypertension (TIPS) is undertaken, or until an acute process (SBP, gastrointestinal bleeding) has been reversed (Table 92-4).93,94
Table 92-4 Management of Hepatorenal Syndrome
ACEIs, angiotensin-converting enzyme inhibitors; ARBs, angiotensin receptor blockers; IV, intravenous; MAP, mean arterial pressure; NSAIDs, nonsteroidal anti-inflammatory drugs; SBP, spontaneous bacterial peritonitis.
* Not available in the United States.
Data from references 69,74,85,86,89,91,93–94,97–124.
Medical Therapy
Terlipressin is an intravenously administered selective vasopressin 1 receptor agonist vasoconstrictor that has been used in Europe and is under review by the FDA for the treatment of type 1 HRS.95,96 It has been evaluated in approximately 330 patients included in four randomized, controlled trials and two meta-analyses.69,97–109 In two multicenter studies of patients with type 1 HRS, terlipressin (given in two different doses in the two studies) in combination with albumin improved serum creatinine levels relative to albumin alone (30% to 43% vs. 8% to 13%), although survival was not significantly different in the two groups.106,107 In addition, in one study,106 terlipressin was associated with a significantly increased rate of cardiovascular complications compared with albumin alone; therefore, this agent should only be used with close monitoring. Finally, the response to terlipressin in patients with type 1 HRS appeared to be better in patients with less severe renal dysfunction at baseline, thus supporting the early initiation of therapy. These studies indicate that administration of terlipressin in combination with albumin can improve renal function in HRS, although the optimal dose and duration of therapy are not defined.
Midodrine, an orally administered α1-adrenergic agonist, and octreotide, a somatostatin analog that inhibits endogenous vasodilators,110,111 have been used in combination with albumin for type 1 HRS in three small nonrandomized studies. In two studies, treatment with midodrine, titrated to cause a rise in mean arterial blood pressure, was associated with improved serum creatinine levels and improved survival compared with no treatment and was associated with few major side effects.112–114 This regimen has the advantage of ease of administration and appears to have a favorable safety profile; however, its efficacy has not been established in randomized controlled trials.
Norepinephrine, a widely available intravenously administered α1-adrenergic agonist, in combination with albumin, has been proposed as an alternative to terlipressin on the basis of two small pilot studies.115,116 In one study of 22 patients with type 1 or 2 HRS, norepinephrine appeared to be as effective and safe as terlipressin. Significant cardiovascular side effects, however, have been reported with the use of norepinephrine to treat HRS, and whether the efficacy and safety of norepinephrine are similar to those of terlipressin have not been fully defined.
Radiologic and Surgical Therapy
Transjugular Intrahepatic Portosystemic Shunt
Insertion of a transjugular intrahepatic portosystemic shunt (TIPS) creates a portosystemic shunt and lowers portal venous pressure, thereby decreasing venous pooling in the splanchnic circulation and increasing venous return. TIPS is effective for the treatment of diuretic-resistant ascites, a precursor to type 2 HRS.117 Four pilot studies have evaluated the use of TIPS in nontransplant candidates with type 1 or 2 HRS.118–121 In these studies, serum creatinine levels declined, sodium excretion increased, and neurohumoral responses improved after TIPS, although survival was not affected. The major benefit was seen in patients with type 2 HRS. In one study, the use of midodrine, octreotide, and albumin followed by TIPS appeared to be effective in a small cohort of patients with type 1 HRS. An important limitation of the use of TIPS in HRS is the potential to worsen hepatic function. Therefore, important questions regarding the safety and benefit of TIPS in HRS remain.
Liver Transplantation
Liver transplantation is the only therapeutic modality that has the potential to reverse both liver dysfunction and HRS and should be considered in any patient found to have HRS.65,66,71,122 Rates of postoperative complications and in-hospital mortality are higher in patients transplanted with HRS than in those without HRS,75,122 and up to 35% of patients with HRS require long-term renal replacement therapy.66 Still, the three-year survival rate of patients transplanted with HRS is approximately 60% compared with 70% to 80% for patients transplanted without HRS. The duration and degree of renal dysfunction preoperatively may be independent predictors of survival, and patients who require hemodialysis carry a mortality risk that is 1.77 times higher than that of patients who do not need dialysis.122–124 In one study, patients with HRS who responded to treatment with a vasopressin analog prior to liver transplantation had outcomes similar to those of patients who underwent liver transplantation without HRS,94 a finding that supports the use of such therapy as a bridge to liver transplantation. Larger trials are needed to confirm this observation.
Other Therapies
Extracorporeal albumin dialysis with MARS is an experimental therapeutic modality that enhances the removal of water-soluble and albumin-binding toxins from the circulation.125 The results of one small randomized trial have supported the use of MARS to improve serum creatinine levels and survival rates in patients with HRS, although larger studies are needed to confirm these findings.126,127 Several other vasoconstrictive medical therapies, including dopamine and octreotide in combination with albumin, have not improved the outcome of HRS, and in one study use of a nonselective endothelin receptor antagonist to inhibit intrarenal vasoconstriction in HRS proved deleterious.95
HEPATOPULMONARY SYNDROME AND PORTOPULMONARY HYPERTENSION
Cirrhosis and portal hypertension are accompanied by alterations in the vascular beds of multiple organ systems. In the pulmonary circulation, two distinct clinical entities, termed the hepatopulmonary syndrome (HPS) and portopulmonary hypertension (PPH), have been recognized. HPS occurs when pulmonary microvascular alterations impair gas exchange and is found in up to 30% of patients evaluated for liver transplantation.128–130 PPH occurs when vasoconstriction and remodeling in resistance vessels increase pulmonary arterial pressures and is found in as many as 5% of patients with cirrhosis. The mechanisms whereby these two entities develop are incompletely characterized, although they occur in similar clinical settings and may share pathogenic pathways. The presence of HPS or PPH increases mortality in affected patients. No effective medical therapies are available for HPS, although liver transplantation reverses the syndrome in most patients. Medical therapies that improve pulmonary hemodynamics in patients with PPH have become available, but the specific role of liver transplantation in PPH is not clearly defined.131–138
PATHOPHYSIOLOGY
Hepatopulmonary Syndrome
HPS is characterized by microvascular alterations and dilatation in the precapillary and capillary pulmonary arterial circulation. In human HPS, the production of vasodilatory substances within the pulmonary vasculature, most notably NO, is increased. Although increased circulating and pulmonary NO levels appear to be features of human HPS, improvement in oxygenation in response to acute inhibition of NO is variable,139–142 and HPS may take more than one year to resolve after liver transplantation in advanced cases.135 These findings suggest that other vasoactive mediators or angiogenesis in the pulmonary microvasculature may contribute to vascular alterations and hypoxemia in human HPS.
In experimental HPS induced by bile duct ligation in the rat, pulmonary NO overproduction has also been observed and is triggered by a series of pathophysiologic events. During the onset of pulmonary vascular alterations, increased biliary production and release of endothelin-1143 in conjunction with shear stress induces pulmonary microvascular endothelin-B receptor overexpression, which leads in turn to endothelin-1–mediated endothelial NO synthase (eNOS)-derived NO production.144,145 As HPS progresses, tumor necrosis factor-α (TNF-α) levels rise as a result of bacterial translocation, which leads to adherence of macrophages in the pulmonary microvasculature and inducible NO synthase (iNOS)–derived NO production and heme oxygenase-1–derived carbon monoxide production.146–150 Endothelin receptor antagonists and inhibition of NOS, bacterial translocation, TNF-α, and heme oxygenase all improve experimental HPS.148–151
Studies have found that experimental HPS is accompanied by enhanced pulmonary vascular endothelial growth factor (VEGF) expression and angiogenesis.151 Both antiangiogenic peptides and pentoxifylline, a phosphodiesterase inhibitor with TNF-α antagonist properties, have been shown to inhibit angiogenesis and decrease the severity of HPS. Two small studies of pentoxifylline in human HPS have reported conflicting results, and the role of angiogenesis and angiogenesis inhibition in human HPS has yet to be defined (Fig. 92-3).152–155
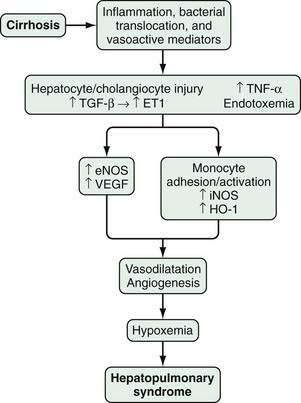
Portopulmonary Hypertension
The mechanisms whereby PPH develops are poorly understood. Histologically, PPH shares the characteristic features of other forms of pulmonary arterial hypertension (PAH): medial proliferation and hypertrophy, plexiform arteriopathy, and in situ vascular thrombosis.156–158 The precise role of portal hypertension in this process is not clear, however, and whether PPH shares pathophysiologic mechanisms with PAH is unknown. Studies have found PPH, like PAH, to be more common in women than men.159






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


