Pathophysiology of Disordered Erythropoiesis in Renal Transplantation
Normal erythropoiesis requires the normal production and secretion of erythropoietin (EPO), adequate levels of iron stores and coenzymes (i.e., folic acid, vitamins B6 and B12, L-carnitine), a normal number of early and late erythroid progenitors (i.e., colony forming units-erythroid [CFU-E] and burst forming units-erythroid [BFU-E]), and a normal red blood cell (RBC) life span.
Erythropoiesis and Production of EPO by the Renal Allograft
Successful renal transplantation results in the normalization of renal function and resolution of anemia of CKD. In contrast to nonrenal forms of anemia in which serum EPO levels are inversely correlated with the levels of Hb, with anemia of renal disease, the lower the EPO levels, the worse the anemia. Therefore, the early resolution of anemia posttransplantation is because of the normal mechanisms that regulate the production and secretion of EPO. The events that result in the normalization of erythropoiesis and the early recovery of anemia can be summarized as follows (
Fig. 21.1) (
25,
26,
27,
28):
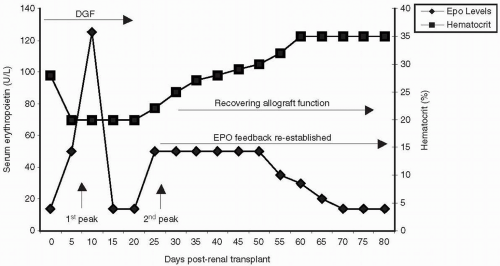
An early rise of EPO secretion starts within the first 24 hours after transplantation (
14,
25,
26). This initial peak is transient and ineffective in generating normal erythropoiesis.
After the first week posttransplant, a smaller and more sustained peak of EPO production occurs. This second peak is associated with the subsequent onset of erythropoiesis and recovery of anemia over the next 2 to 3 months (
29,
30,
31).
By 10 weeks after transplant, serum EPO concentrations should reach 100% of the expected levels in individuals with normal functioning grafts (
32).
Early Posttransplant Anemia
The natural recovery of EPO production by the functional allograft is disrupted by early allograft injury such as acute rejection or ischemia reperfusion. The onset of acute rejection within the first month after renal transplantation delays the second peak of erythropoietic response until after the successful resolution of rejection (
29). Acute rejection affects the recovery of erythropoiesis by mechanisms other than the generation of allograft dysfunction, such as the generation of a proinflammatory environment (
33,
34).
Rejection can also increase the severity of anemia if antilymphocyte antibody (Ab) therapy is used. Polyclonal and monoclonal Ab preparations are known to shorten RBC lifespan due to anti-RBC heterophile Ab-induced hemolysis or hemolytic uremic syndrome-thrombotic thrombocytopenic purpura (HUS-TTP)-like syndrome (
35,
36). However, polyclonal and monoclonal Ab’s are not directly toxic to erythroid precursors, and are effective treatments for immune aplastic anemias and clonal disorders such as myelodysplastic syndrome and paroxysmal nocturnal hemoglobinuria (PNH) (
37,
38,
39,
40).
Ischemic injury and other causes of delayed graft function (DGF) are important etiologies of inadequate erythropoiesis in renal allograft recipients. Both the resumption of EPO secretion and consequent reticulocyte response do not occur unless the allograft is fairly functional. In contrast, patients with DGF or slow graft function achieve only 75% of their expected concentrations of EPO, remain significantly anemic and take longer to develop a normal erythrocytosis (
32). In recipients of expanded criteria donor renal allografts, erythropoiesis may not be restored to normal levels. These patients are also at increased risk for acute rejection, and thus worse anemia.
Late Posttransplant Anemia
To date, few studies have rigorously studied the pathogenesis and natural history of late PTA (
3,
41). There appear to be decreased numbers of CFUs-E with normal to increased numbers of BFUs-E, with high burst promoting activity. Level of EPO is also disproportionately low to the degree of chronic allograft dysfunction and the decrease in the observed/expected EPO levels ratio. A disruption of the EPO regulatory feedback is likely an important pathogenic role. Nevertheless, this should not discourage the use of rHuEPO in these patients, as the reduced CFU-E responds to adequate doses of the hormone (
42).
Posttransplant Erythrocytosis
The pathophysiology of PTE is heterogeneous (
16), and both EPO-dependent and independent mechanisms have been implicated in its pathogenesis. In patients with EPO-dependent PTE, both a deficient feedback regulation of erythropoiesis (
43) and increased levels of EPO have been described (
42). The persistent secretion of EPO by ischemic and diseased native kidneys appears to play the primary role (
13). In patients with EPO-independent PTE, BFUs-E display high sensitivity to reduced doses of EPO
in vitro (
44), and in some cases, spontaneous EPO-independent growth of BFU-E and CFU-E has been documented (
16). In this subgroup of patients, the action of other growth factors such as angiotensin II (Ang II), androgens and insulin-like growth factor-1 (IGF-1) on erythroid progenitors are important stimulants (
13,
45). In erythroid progenitors, Ang II affects erythropoiesis by binding to its Ang II-receptor 1 (AT1) and activating JAK2 kinase required for EPO-dependent erythropoiesis (
46,
47,
48). Androgens promote erythropoiesis by the direct stimulation of erythroid progenitors, the stimulation of endogenous EPO secretion or via the activation of Ang II (
49,
50,
51). IGF-1 is a regulator of erythropoiesis
in vivo (
52,
53,
54) and has been identified as the major circulating factor supporting erythropoiesis in anephric dialysis patients with no measurable EPO levels (
55). IGF-1 binding proteins 1 and 3 (IGFBP-1 and IGFBP-3) modify IGF-1 function by facilitating the interaction with its receptor (
53,
54). Patients with PTE, when compared to normal individuals or renal transplant patients with normal Hct, have a significant increase in the serum level of IGF-1, IGFBP1, and IGFBP3 (
52). Moreover, the effectiveness of ACEI in the treatment of PTE has been attributed in part to their ability to decrease IGF-1 levels (
56,
57).
Iron and Coenzyme Metabolism after Renal Transplantation
Normal iron homeostasis is necessary for the maintenance of normal erythropoiesis. Iron deficiency is a common problem in renal transplantation with up to 50% of allograft recipients reported to be iron deficient by day 14 after renal transplantation (
58). Relative or absolute iron deficiency is multifactorial in renal transplant recipients, possible etiologies including pretransplant iron deficiency, gastrointestinal
blood loss, frequent blood sampling, a proinflammatory environment and prior rHuEPO use with subsequent high iron demands. Serum ferritin decreases in the early posttransplant period and reaches normal levels by 36 months after successful transplantation. About one third of renal transplant patients develop persistently low serum ferritin levels (
50). Deficiency of coenzymes (i.e., folate, vitamin B6, vitamin B12 and vitamin C) can result in nuclear maturation defects of erythroid progenitors and resistance to EPO. In patients with acute rejection and anemia, there is a downregulation of genes for folate necessary for the procurement of folate, resulting in a reduced ability to transport folate and a relative deficiency that can be overcome with adequate supplementation (
33).
Malignancy, Infection and Medications Causing Defective Erythropoiesis after Renal Transplantation
PTA can be a cardinal sign of malignancy in patients with renal transplantation. Infection has a significant impact on the occurrence of anemia after renal transplantation (
2). In renal transplant recipients, acute viral infections and reactivation of viral infection have a greater impact on the occurrence of anemia than chronic viral infections (
2). There is a long list of viruses that can cause PTA, including cytomegalovirus (CMV), Epstein-Barr virus (EBV), human herpes virus (HHV)-6, HHV-8, herpes simplex virus (HSV), hepatitis B and C, and rubella (
59,
60). Of note is parvovirus B-19, which can cause a pure red cell aplasia as well as the HUS-TTP syndrome (
61,
62,
63,
64,
65). There are a number of potential mechanisms, including direct infection by parvovirus of CFU-E and BFU-E, autoantibody production, and induction of autoimmune destruction by T cells (
66). Diagnosis of active infection is best made by nested polymerase chain reaction (PCR). Treatment of parvovirus B-19 includes a reduction in immunosuppression and intravenous immunoglobulin (IVIG) (
62,
67).
Medications are among the most common causes of defective erythropoiesis in transplanted patients and the general population. With the exception of corticosteroids, all immunosuppressants can cause anemia by any of the three mechanisms discussed above. Drug-induced anemias can be classified in three groups according to their pathogenesis:
Drug-induced maturation disorders: Maturation disorders are caused primarily by drug-induced disruption of DNA synthesis in the nucleus of erythroid precursors, or by drug-induced cytoplasmic abnormalities of mitochondrial function resulting in defective heme synthesis. Antiproliferative agents (i.e., AZA, MMF, rapamycin) and drugs that interfere with absorption or metabolism of coenzymes (i.e., AZA, MMF and anticonvulsant agents) are known causes of anemias due to nuclear maturation disorders. This form of anemia is characterized by the presence of macrocytosis with or without hypochromia. Anticonvulsants and other agents of drug-induced porphyrias disrupt the cytoplasmic maturation of erythroid precursors by interfering with mitochondrial pathways that regulate the incorporation of iron into the heme molecule. Anemias resulting from these defects have microcytic and hypochromic morphology.
Drug-induced bone marrow hypoproliferation: Drugs decrease bone marrow proliferation by a selective arrest of the cell cycle of CFU-E and BFU-E (a pure RBC aplasia). Pure RBC aplasias (PRCAs) can be due to direct toxicity to erythroid progenitors by the drug or its metabolites, or immune mechanisms such as auto-Ab, T-cell-mediated or natural killer cell-mediated injury. In addition, drugs can induce chromosomal abnormalities and erythroid clonal defects that result in myelodysplasia-like disorders. ACEIs and angiotensin II receptor 1 blockers (ARBs) (
68,
69,
70,
71) can cause hypoproliferative anemias through mechanisms different to the ones listed above. By inhibiting IGF-1 production (
69) and increasing the half-life and serum levels of AcSDKP, a natural peptide that inhibits erythropoiesis and which hydrolysis is dependent on ACE activity (
70,
72), ARBs and ACEIs can induce decreased proliferation of erythroid progenitors.
Drug-induced hemolysis: Drug-induced hemolysis can be classified into two large groups: (a) Intracorpuscular: if the mechanism responsible for hemolysis is intrinsic to the RBC, such as with enzymatic or membrane defects. An example of drug-induced intracorpuscular hemolytic anemia (DIHA) is deficiency of glucose-6-phosphate dehydrogenase (G6PD). This is the most common cause of DIHA in susceptible renal transplant patients. The highest risk patient populations are those of African and Mediterranean ancestry. G6PD catalyses the first step in the pentose phosphate pathway that results in the reduction of NADP
+ to NADPH, which decreases the susceptibility of the RBC to drug-induced oxidative damage and the apoptosis of erythroid precursors (
73,
74). Sulfacontaining antibiotics, dapsone and antimalarials are the most common causes of DIHA in G6PD-deficient individuals. (b) Extracorpuscular: if the mechanism responsible for hemolysis is extrinsic to the RBC, as it happens in autoimmune hemolytic anemia (AIHA) where Ab-mediated injury occurs, or microangiopathic anemia (HUS-TTP-like disorders) where the pathogenesis involves endothelial cell injury. Drug-induced extracorpuscular hemolytic anemias (DEHA) are Ab-mediated. All forms of DEHA are elicited by IgG or warm Ab, and are direct Coombs test positive. However, the target of the Ab response varies from the Rh group of RBC antigens in the auto-Ab forms to the drug-RBC glycoprotein complex in the hapten forms.
Diagnosis and Treatment of Posttransplant Anemia
The diagnostic work-up of PTA is similar to the approach for the nontransplant patient but should be modified to recognize specific etiologies of anemia in the renal transplant population. Initial PTA evaluation should include (a) Hb
measurements, (b) evaluation of RBC morphology (c) evaluation of iron stores, (d) evaluation of coenzyme levels (in certain subpopulations), (e) evaluation of erythropoiesis, and (f) evaluation for occult blood loss (
17). These parameters are helpful to detect the cause of many anemias that are not due to EPO deficiency (
75). Although EPO levels have been measured for the evaluation of erythropoiesis in renal transplantation (
25,
26,
29,
30), its use is primarily academic and should not be part of the initial approach to PTA (
17). Anemia in renal transplant recipients with serum creatinine ≥2 mg/dL likely is due to EPO deficiency; nevertheless, other etiologies should also be excluded. Bone marrow aspirates and biopsies are usually reserved for the evaluation of early or late PTA with hypoproliferative features.
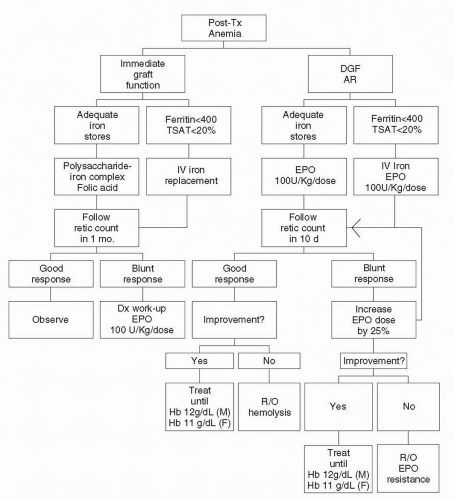
Assessment of RBC morphology requires the evaluation of RBC indices and a peripheral blood smear. Posttransplant anemia, similar to anemia of CKD, is normocytic and normochromic. The presence of low or high mean corpuscular volume (MCV), spherocytes or schistocytes should prompt immediate additional work-up as proposed in
Figure 21.2. Hb rather than Hct is increasingly being used to quantify the degree of anemia (
76,
77,
78).
The basic evaluation of iron stores should include the measurement of ferritin and transferrin saturation levels (TSAT). Although well defined for the management of anemia of CKD, guidelines for the evaluation of iron stores have not been established in renal transplant recipients. Serum ferritin is no longer considered the best parameter for the assessment
of iron stores. Until more accurate indicators such as free erythrocyte protoporphyrin or the reticulocyte maturation index (RMI) become readily available, clinicians will continue to rely on serum ferritin and TSAT (
14). All recipients should undergo the evaluation of iron stores and bone marrow erythropoiesis early after transplantation, including measuring serum ferritin level, TSAT and reticulocyte count.
Serum soluble transferrin receptor levels (sTRF-R) and the RMI have been reported to be sensitive early predictors of bone marrow responsiveness and erythropoiesis following renal transplantation (
79,
80,
81,
82). Neither test, however, is readily available in routine clinical practice, leaving the absolute and relative reticulocyte counts as the only practical tests to evaluate the bone marrow response to endogenous and exogenous EPO. The reticulocyte count, a representation of graft EPO production, allows renal transplant patients to be divided in two groups. The first group are patients who have immediate allograft function and develop a reticulocyte peak before day 29 posttransplant. In these patients the resolution of early PTA occurs within 6 weeks provided that iron stores are available and no rejection occurs. The second group are patients with DGF or intermediate graft function who do not develop a reticulocyte peak before day 29 and frequently develop early and late PTA (
83). Patients in the second group should receive rHuEPO therapy.
The evaluation of blood loss in renal transplant recipients requires adequate communication with the whole transplant team to minimize insensible blood loss in the form of frequent and large volume phlebotomy. In addition, gastrointestinal (GI) blood loss must be excluded. The testing of stool for occult blood is usually sufficient to exclude GI blood loss as a cause of refractory anemia, though occasionally endoscopic studies are necessary.
Replacement of depleted iron stores and EPO levels are necessary for the correction of PTA. Oral iron is poorly absorbed, often not tolerated, and can interfere with the absorption of immunosuppressants. Thus our recommendation for replenishment of iron stores is intravenous iron sodium gluconate (
84). For patients with adequate iron stores and normal allograft function, we recommend an oral polysaccharide-iron complex with folate. Treatment is usually discontinued within the first 3 months posttransplant, once an Hb level of 11 g/dL is achieved. In all patients with DGF, intermediate graft function or acute rejection, rHuEPO should be administered at an average dose of 100 U/kg SQ at least weekly. As EPO resistance is common in these patients (
Table 21.1), if a reticulocyte spike is not detected 7 days posttransplantation, the dose of rHuEPO is increased by 25%. These patients should also undergo evaluation for secondary or tertiary hyperparathyroidism, hypothyroidism, hemolysis, and GI blood loss as cause of EPO resistance.
Many have advocated the replacement of coenzymes as an adjuvant therapy to rHuEPO in the management of anemia of CKD (
19,
85,
86). The benefit of these interventions has not been tested in renal transplant recipients. Vitamin B12 deficiency should be excluded in all patients with megaloblastic features with or without anemia because folate replacement can easily mask the classical features of pernicious anemia. The use of vitamin B6 and ascorbate is reserved for the treatment of hypoproliferative early or late PTA with adequate iron stores and persistent microcytosis (
86) or renal transplant recipients with hemoglobinopathies (
87).
Diagnosis and Treatment of Posttransplant Erythrocytosis
All renal transplant recipients with an Hb level >17-18 g/dL for more than 3 months without evidence of volume contraction or rHuEPO replacement should be evaluated for PTE. As in the general population, primary polycythemia
(polycythemia rubra vera [PRV]) and overlapping myeloproliferative syndromes should be excluded. Secondary forms of polycythemia such as EPO producing tumors (i.e., hepatocellular and renal cell carcinomas, uterine leiomyomata), chronic viral hepatitis (i.e., hepatitis B or C), chronic obstructive pulmonary disease, pulmonary hypertension or intracardiac shunt with longstanding hypoxemia should be excluded. Although the diagnosis of true polycythemia requires isotopic determination of RBC mass and plasma volume, this test is seldom used. The basic evaluation for PTE includes: (a) EPO levels, (b) measurement of the partial arterial pressure of oxygen (PaO
2) by arterial blood gases, (c) imaging of the abdomen and pelvis, and (d) liver function tests and viral hepatitis serologies.
In patients with low EPO levels, the differential diagnosis is limited to PRV or PTE. In these patients, elevated vitamin B12 and leukocyte alkaline phosphatase levels would confirm the diagnosis of PVR (
88). In patients with high EPO levels and low PaO
2, the differential diagnoses include chronic pulmonary diseases, smokers’ polycythemia or intracardiac shunts. A high carboxyhemoglobin level would confirm smokers’ polycythemia. If the carboxyhemoglobin level is low, an extrarenal source of EPO production should be sought. In this last category, malignant tumors that produce EPO and hepatocellular disease should be excluded.
Since the recognition that ACEI and ARBs reduce the Hb levels in hemodialysis and renal transplant patients, these drugs have become the cornerstone in the management of PTE, relegating phlebotomy and theophylline to second line therapies (
89,
90). The doses of ACEI and ARBs required to achieve a decrease in Hb do not differ from the standard doses used for the treatment of hypertension. As PTE patients are also often hypertensive, the treatment is well tolerated. Five to 10% of patients with PTE are resistant to ACEI or ARBs (
68) yet, in select patients, a combination of an ACEI and theophylline may be effective therapy (
91). Otherwise, phlebotomy remains the only effective therapy and should be carried out if the Hb level reaches 18 g/dL (
92).
 Detection of Recipient Pretransplant Alloreactivity
Detection of Recipient Pretransplant Alloreactivity
 Surgical Issues in the Transplant Recipient
Surgical Issues in the Transplant Recipient
 Posttransplantation Liver Disease
Posttransplantation Liver Disease
 Cytomegalovirus in Renal Transplantation
Cytomegalovirus in Renal Transplantation
 Medical Complications of the Eyes, Nasopharynx, Dentition, Oropharynx, and Hearing in the Kidney Transplant Recipient
Medical Complications of the Eyes, Nasopharynx, Dentition, Oropharynx, and Hearing in the Kidney Transplant Recipient



