(1)
Division of Nephrology and Hypertension, Rutgers New Jersey Medical School, Newark, NJ, USA
1.
A 20-year-old athletic man with no significant past medical history is found to have isolated asymptomatic microscopic hematuria on routine phys exam . He is not on any medications, and does not use illicit drugs. Which one of the following observations suggests hematuria of glomerular origin ?
A.
Presence of many isomorphic red blood cells (RBCs) in urine sediment
B.
Presence of many isomorphic RBCs and white blood cells (WBCs) in urine sediment
C.
Presence of many dysmorphic RBCs or acanthocytes in urine sediment
D.
Presence of many “decoy cells” in urine sediment
E.
All of the above
The answer is C
Urinary RBCs or erythrocytes are of two types: isomorphic and dysmorphic. Isomorphic RBCs have regular shapes and contours. These RBCs generally originate from lower urinary tract. Dysmorphic RBCs have irregular shapes and contours, and originate from renal parenchyma (glomeruli). Thus, hematuria is considered either glomerular or nonglomerular in origin . Hematuria is also seen in several nonrenal conditions such as exercise, fever, and/or menstruation.
Glomerular hematuria is considered when ≥40 % dysmorphic RBCs or 5 % acanthocytes or RBC casts are present. Dysmorphic RBCs are best visualized by phase-contrast microscopy.
RBCs acquire dysmorphism while they are passing through gaps of the glomerular basement membrane . Also, physicochemical insults occur when RBCs pass through the renal tubules. It has been shown that the number of dysmorphic RBCs is higher in proliferative glomerulonephritis than in nonproliferative glomerulonephritis.
The presence of “decoy cells,” in the urine is from a virus called BK polyomavirus . It is a double-stranded DNA virus that affects about 90 % of the general population. Interestingly, a number of renal transplant patients develop renal as well as graft failure due to infection of this virus. The “decoy cells” bear viral inclusion bodies, and their presence in the urine indicates the infection with BK virus. Decoy cells show glossy-appearing intranuclear viral inclusion bodies mostly coming from the uroepithelium.
In this individual, there is no evidence of infection or renal disease. His hematuria can be attributed to exercise. Thus, options A, B, D, and E are incorrect.
Suggested Reading
Fogazzi GB, Edfonti A, Garigali G, et al. Urinary erythrocyte morphology in patients with microscopic hematuria caused by a glomerulopathy. Pediatr Nephrol 23:1093–1110, 2008.
Fogazzi GB. Urinalysis. In Floege J, Johnson RJ, Feehally J (eds). Comprehensive Clinical Nephrology, 4th ed, Philadelphia, Saunders/Elsevier, 2010, pp. 39–55.
2.
An 18-year-old adult is found to have persistent asymptomatic isolated microscopic hematuria (PAIMH) >5 RBCs/hpf during phys exam for military service. There is no significant personal or family history of renal or urologic disease. He is not on any medications. Phys exam, blood pressure, and serum chemistry are normal. There is no proteinuria on urine dipstick . In view of recent evidence, which one of the following statements is CORRECT?
A.
He is at risk for progressive renal disease
B.
He warrants testing for genetic disease
C.
He is not at risk for any kidney disease
D.
He is at low risk for future kidney disease and needs follow-up
E.
He does not need any follow-up and ignore hematuria
The answer is D
PAIMH, once thought to be a benign disease, seems to carry a low risk for future development of either hereditary nephritis, thin basement membrane disease , IgA nephropathy , or other diseases with progression to end-stage renal disease ( ESRD) . In a retrospective study of Vivante et al. from Israel, 0.3 % (3690 out of 1,203,626 subjects) of young adults aged 16–25 years were found to have PAIMH. At a follow-up of 22 years, 26 of 3690 (0.7 %) participants with PAIMH developed treated ESRD, compared to only 0.045 % of participants without hematuria. A multivariate analysis showed that the hazard ratio for treated ESRD with hematuria versus non hematuria was 18.5 %. However, the absolute risk of progression is low (3 ESRD/1000). Thus, PAIMH carries a low absolute risk for ESRD in Israeli young adults. Whether or not PAIMH carries such a risk for ESRD in other populations remains to be seen. Thus, option D is correct.
Suggested Reading
Kovacević Z, Jovanović D, Rabrenović V, et al. Asymptomatic microscopic hematuria in young males. Int J Clin Pract 62:462–412, 2008.
Vivante A, Afek A, Frenkel-Nir Y, et al. Persistent asymptomatic isolated microscopic hematuria in Israeli adolescents and young adults and risk for end-stage renal disease. JAMA 306:729–736, 2011.
3.
A 16-year-old adolescent is found to have orthostatic proteinuria (1.2 g/day). Which one of the following you suggest regarding his risk for future development of renal disease?
A.
High risk and treatment is required
B.
Low risk and no treatment is required
C.
No risk and no treatment is required
D.
Renal biopsy is required to assess risk
E.
Repeat proteinuria is required in 2 weeks to assess risk
The answer is C
Orthostatic proteinuria is common in children and adolescents. It is uncommon in individuals older than 30 years of age. Orthostatic proteinuria is defined as the presence of proteinuria (up to 2 g) in upright position and very little proteinuria (<50 mg) in supine position. The pathogenesis of proteinuria is unclear, however, renal hemodynamic changes and entrapment (kinking) of left renal vein between aorta and superior mesenteric artery have been implicated. Long-term (20 year) follow-up of subjects with orthostatic proteinuria showed no progression of renal disease. Proteinuria resolves in 50 % of subjects after 10 years and 87 % after 20 years. Therefore, orthostatic proteinuria is a benign condition, and no treatment or renal biopsy is required. Thus, option C is correct.
Suggested Reading
Springberg PD, Garrett Jr LE, Thompson Jr A.L, et al. Fixed and reproducible orthostatic proteinuria: results of a 20-year follow-up study. Ann Intern Med 97:516–519, 1982.
Shintaku N, Takahashi Y, Akaishi K, et al. Entrapment of left renal vein in children with orthostatic proteinuria. Pediatr Nephrol 4:324–327, 1990.
Devarajan P. Mechanisms of orthostatic proteinuria: lessons from a transplant donor. J Am Soc Nephrol 4:36–39, 1993.
4.
A 30-year-old Caucasian man is referred to you by a primary care physician for evaluation of 2+ (100 mg/dL) proteinuria confirmed on three different visits on a routine urinalysis. There is no hematuria. The personal and family history is unremarkable. He is not on any medications. A 24 h proteinuria is 1.2 g. Blood pressure is normal. Serum creatinine is 0.8 mg/dL with an estimated glomerular filtration rate (eGFR) >60 mL/min/1.73 m2. Regarding the status of his renal function in the next 7–10 years, which one of the following statements is CORRECT?
A.
Difficult to predict at this time
B.
Renal biopsy is required to evaluate renal function
C.
No relationship exists between isolated proteinuria and renal function
D.
Annual estimation of eGFR is required to evaluate renal function status
E.
There is a possibility that his eGFR may decline >5 %/year from baseline in the next 7 years
The answer is E
Persistent isolated proteinuria has been found to be strongly associated with progression of kidney disease, and cardiovascular as well as all-cause mortality. In population studies, dipstick proteinuria was identified as an important predictor of ESRD. Also, routine testing for proteinuria using dipstick can predict future renal function decline.
Clark et al. evaluated the predictive value of dipstick proteinuria of different levels for rapid kidney function decline (RKFD) in 2574 Canadian participants (18–92 years of age) over a median of 7 years follow-up. RKFD was defined as a >5 % annual eGFR change from baseline. Overall, 2.5 % ( N = 63) had ≥100 mg/dL (≥1 g/L) or ≥2+ proteinuria. Of these, 8.5 % experienced RKFD during a 7-year follow-up. One in 2.6 patients with proteinuria >100 mg/dL experienced RKFD. Participants >60 years of age and eGFR <60 mL/min were at higher risk for RKFD. Dipstick proteinuria >100 mg/dL correctly identified RKFD in 91 %, incorrectly in 1.7 % and missed RKFD in 7.7 % of participants.
In this study, proteinuria was found to be a strong predictor for RKFD than albuminuria. After adjusting for age, hypertension, diabetes, cardiovascular disease, obesity, and family history of diabetes were all significantly associated with increased risk for RKFD. The detection of trace protein or greater had more than two-fold increase in the risk of RKFD. Thus, inexpensive dipstick screening for proteinuria can detect those individuals at risk for RKFD . Of all the options, option E is, therefore, correct.
Suggested Reading
Iseki K, Iseki C, Ikemiya Y, et al. Risk of developing end-stage renal disease in a cohort of mass screening. Kidney Int 49:800–805, 1996.
Ishani A, Grandits GA, Grim RH, et al. Association of single measurements of dipstick proteinuria, estimated glomerular filtration rate, and hematocrit with 25-year incidence of end-stage renal disease in the multiple risk factor intervention trial. J Am Soc Nephrol 17:1444–1452, 2006.
Clark WF, Macnab JJ, Sontrop JM, et al. Dipstick proteinuria as a screening strategy to identify rapid renal decline. J Am Soc Nephrol 22:1729–1736, 2011.
5.
A-35-year-old woman was found to have asymptomatic hematuria with albumin:creatinine ratio <0.02 mg/mg. Which one of the following diagnoses is MOST likely on a renal biops y?
A.
IgA nephropathy
B.
Alport syndrome
C.
Thin basement membrane disease (TBD)
D.
MPGN (type I)
E.
Lupus nephritis (Class I)
The answer is C
In a study of Hall et al. patients with asymptomatic microscopic hematuria and normal albumin excretion rate (<20 mg/day or albumin:creatinine ratio (ACR) <0.02 mg/mg or <30 mg/day) were found to have mostly TBD (43.1 %). About 20.1 % had IgA nephropathy, 20.1 % had nondiagnostic abnormalities , and 18.1 % had normal findings.
Other studies have shown that 50–90 % of patients with microalbuminuria (ACR >0.02 mg/mg or >30 mg/day) and microscopic hematuria were found to have IgA nephropathy. On the other hand, patients with hematuria with normal albumin excretion rate were found to have mostly TBD. Shen et al. reported in Chinese adult subjects with isolated microscopic hematuria and elevated serum IgA/C3 ratio (~4.5) and an ACR of 96 mg/g are more common in IgA nephropathy than subjects with TBD. Thus, the degree of albuminuria in a patient with microscopic hematuria can predict the type of renal disease on a renal biopsy .
Suggested Reading
Eardley KS, Ferreira MA, Howie AJ, et al. Urinary albumin excretion: A predictor of glomerular findings in adults with microscopic hematuria. QJM 97:297–301, 2004.
Hall CL, Bradley R, Kerr A, et al. Clinical value of renal biopsy in patients with asymptomatic microscopic hematuria with and without low-grade proteinuria. Clin Nephrol 62:267–272, 2004.
Assadi FK. Value of urinary excretion of microalbuminuria in predicting glomerular lesions in children with isolated microscopic hematuria. Pediatr Nephrol 20:1131–1135, 2005.
Shen P, He L, Jiang Y, et al. Useful indicators for performing renal biopsy in adult patients with isolated microscopic hematuria. Int J Clin Pract 61:789–794, 2007.
6.
A 50-year-old housewife visits her physician for extreme wrist and knee pain. She has been taking ibuprofen for a number of years with partial relief of her pain. She denies fatigue, weakness, and/or lower extremity edema. Blood pressure is elevated. Urinalysis shows >300 mg proteinuria with a protein to creatinine ratio of 4.5 (mg/mg). There is no hematuria. Which one of the following glomerular diseases is MOST likely found on a renal biopsy?
A.
Focal segmental glomerulosclerosis (FSGS)
B.
Minimal change disease
C.
Membranoproliferative glomerulonephritis (MPGN)
D.
Amyloidosis
E.
Chronic GN (glomerulonephritis)
The answer is B
The patient has been taking ibuprofen for many years. The ingestion of nonsteroidal anti-inflammatory drugs (NSAIDs) such as ibuprofen can cause proteinuria from non-nephrotic range to nephrotic range. Generally nephrotic syndrome is due to a glomerular lesion , most commonly minimal change disease. Few reports have also described nephrotic syndrome due to membranous nephropathy in NSAID users . FSGS and MPGN have not been described. Amyloidosis with nephrotic syndrome can be seen in patients with rheumatoid arthritis, but the patient does not have any other manifestations of amyloidosis. Chronic GN is a possibility, but the absence of hematuria rules out this disease. Also, nephrotic syndrome is less likely in chronic GN. Thus, option B is correct.
Suggested Reading
Kirschenbaum MA, Shah GM. Nephropathy of nonsteroidal anti-inflammatory agents. In Massary SG, Glassock RJ (eds) Massry & Glassock’s Textbook of Nephrology, 3rd ed, Baltimore, Williams & Wilkins, 1995, pp. 940–947.
Palmer BF, Henrich WL. Toxic nephropathy. In Brenner BM (ed): Brenner & Rector’s The Kidney, 7th ed, Philadelphia, Saunders, 2004, pp. 1625–1658.
Palmer BF. Nephrotoxicity of nonsteroidal anti-inflammatory agents, analgesics, and inhibitors of the renin-angiotensin system. In Coffman TM, Falk RJ, Molitoris BA, et al. (eds) Schrier’s Diseases of the kidney 9th ed, Philadelphia, Wolters Kluwer/Lippincott Williams & Wilkins, 2013, pp. 943–958.
7.
A 12-year-old female student with nephrotic syndrome (10 g/day) is seen in renal clinic for management of her proteinuria. She is started on oral prednisone with good response (proteinuria 250 mg/day) in 9 weeks. Prednisone is slowly tapered to 5 mg every other day. She is not hypertensive; however, she is just started on 2.5 mg of enalapril. After 2 years of remission, she presents to the clinic with peripheral edema, and urinary protein excretion increased to 6.5 g/day. Her renal function is normal. Which one of the following urinary findings is SUGGESTIVE of relapse of her nephrotic syndrome ?
A.
Urinary albumin:protein ratio
B.
Urinary neutrophil gelatinase-associated lipocalin (NGAL)
C.
Monocyte chemoattractant protein-1 (MCP-1)
D.
Urinary CTLA-4
E.
Urinary CD80
The answer is E
Garin et al. reported a urine marker for relapse of idiopathic minimal change disease mostly in children . It is named CD80 (also known as B7.1), which is expressed on podocytes in various animal models of nephrotic syndrome . CD80 is a transmembrane protein that provides a co-stimulatory signal for T-cell activation. Its expression is always activation-induced. It is reported that induction of CD80 by podocytes causes proteinuria and fusion of foot processes in mice. Garin et al. have shown that urinary CD80 levels are increased during relapse and normalization during remission in children with minimal change disease. Elevation in urinary CD80 was not observed in other glomerular diseases.
T-regulatory cells also secrete soluble CTLA-4, which binds to CD80 and can block its co-stimulatory activation of T cells. Unlike CD80, the urinary levels of soluble CTLA-4 are not elevated during relapse of minimal change disease. However, the ratio of CD80/CTLA-4 is elevated. Thus, option D is incorrect.
MCP-1 levels are increased in the urine during lupus flare and not in minimal change disease. Urinary NGAL is elevated in AKI prior to an increase in serum creatinine levels . Increased urinary albumin:protein ratio differentiates glomerular from nonglomerular causes of hematuria. Thus, options A to D are incorrect.
Suggested Reading
Ohisa N, Yoshida K, Matsuki R, et al. A comparison of urinary albumin-total protein ratio to phase-contrast microscopic examination of urine sediment for differentiating glomerular and nonglomerular bleeding. Am J Kidney Dis 52:235–241, 2008.
Garin EH, Diaz LN, Mu W, et al. Urinary CD80 excretion increases in idiopathic minimal-change disease. J Am Soc Nephrol 20:260–266. 2009.
8.
An 18-year-old African American man is found to have hematuria and 3+ proteinuria on urinalysis during a routine phys exam. He is referred to a nephrologist for further evaluation. A 24 h urine collection shows 5.2 g of proteinuria. His BP is 148/86 and a pulse rate of 76. Except for trace edema, his phys exam is otherwise normal. Serum chemistry and CBC are normal. Serum complement and ASO titers are normal. ANA and anti-DNA antibodies are negative. A renal biopsy shows the following:
Light microscopy (LM): normal glomerular appearance
Electron microscopy (EM): mesangial immune complex dense deposits
Immunofluorescence (IF) microscopy: 2+ IgG, 2+ IgM, 2+ C3 and 4+ C1q and absent κ and λ chains in glomerular mesangium
Which one of the following choices is the MOST likely diagnosis in this patient?
A.
Minimal change disease
B.
Focal segmental glomerulosclerosis (FSGS)
C.
Lupus nephritis (Class I)
D.
C1q nephropath y
E.
Orthostatic proteinuria
The answer is D
The diagnosis in this young patient is C1q nephropathy, which is a rare renal disease that resembles histologically lupus nephritis . C1q nephropathy is seen predominantly in young (15–30 years of age) subjects with male dominance. African American and Hispanics may have a higher incidence. The clinical manifestations include proteinuria in the nephrotic range or nephrotic syndrome with normal to decreased GFR. Hematuria, edema, and hypertension are present in substantial number of patients. Interestingly, C1q nephropathy is usually diagnosed during a routine phys exam. In these patients, LM findings vary from normal appearing glomeruli (mimicking minimal change disease ) to FSGS . IF and EM show the presence of dominant or co-dominant deposits of C1q with less staining for immunoglobulins and C3 and electron-dense deposits in the mesangial region. The staining for κ and λ chains is absent. Serum complement levels are normal. Renal survival at 3 years of follow-up is 84 %, and proteinuria does not always respond to corticosteroids.
Based on the IF microscopy findings, C1q nephropathy may be confused with class 1 or class 2 lupus nephritis. However, absence of lupus serology and normal serum complement levels, absence of tubuloreticular inclusion bodies, lack of staining for κ and λ chains, and absence of anti-C1q antibodies exclude the diagnosis of lupus nephritis .
The presence of immune deposits excludes the diagnosis of both minimal change disease and FSGS . Orthostatic proteinuria, which is defined as the presence of proteinuria in the upright position and the absence of proteinuria in the supine position, should be considered in any young individual with proteinuria around 1–2 g/day. In these subjects, the renal biopsy is normal and the course of proteinuria is benign. Follow-up in 6–12 months is needed to ascertain that the degree or pattern of proteinuria has not changed, and corticosteroid treatment is not indicated.
Suggested Reading
Sharman A, Furness P, Feehally J. Distinguishing C1q nephropathy from lupus nephritis. Nephrol Dial Transplant 19:1420–1426, 2004.
Vizjak A, Ferluga D, Rozic M, et al. Pathology, clinical presentations, and outcomes of C1q nephropathy. J Am Soc Nephrol 19:2237–2244, 2008.
Nachman PH, Jennette JC, Falk RJ. Primary glomerular diseases. In Taal MW, Chertow GM, Marsden PA, et al. (eds): Brenner & Rector’s The Kidney, 9th ed, Philadelphia, Elsevier Saunders, 2012, pp. 1100–1191.
9.
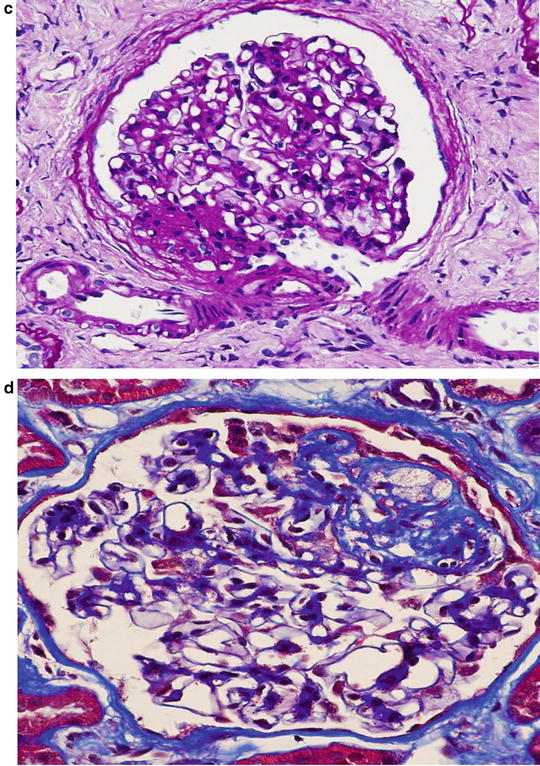
Match the following idiopathic glomerular lesions (variants) of focal segmental glomerulosclerosis (FSGS) with the renal outcomes, as described from 1 to 4
1.
Patients with severe nephrotic syndrome and deteriorating renal function with time
2.
Patients with severe nephrotic syndrome and least impaired renal function with time
3.
Patients with lowest frequency of nephrotic syndrome and highest frequency of hypertension
4.
Similar to perihilar variant with less severe glomerular sclerosis and tubulointerstitial injury
Answers: A = 1; B = 2; C = 3; D = 4
Idiopathic focal segmental glomerulosclerosis (FSGS) has been classified into five variants based on histologic features and renal outcomes. They are: (1) collapsing FSGS; (2) tip lesion FSGS; (3) cellular FSGS; (4) perihilar FSGS; and (5) FSGS not otherwise specified (FSGS NOS). A recent study reviewed the biopsy specimens of 282 patients with 20 years of follow-up, and compared the clinical outcomes and response to treatment among these five variants. Collapsing variant had a striking predilection for African Americans, and patients with this form had severe nephrotic syndrome, substantial renal insufficiency, and poor renal survival. Only 14 % of patients were in complete remission at the end of follow-up. Therefore, Fig. 2.1a is consistent with option 1 (collapsing FSGS).
Tip lesion FSGS was present in low percentage of patients; however, they had severe nephrotic syndrome, but the initial and final serum creatinine levels were least affected. Of all FSGS variants, patients with tip lesion variant had the highest rate of complete remission as well as the highest rate of renal survival. Thus, Fig. 2.1b is compatible with option 2 ( tip lesion FSGS).
Patients with perihilar FSGS had the lowest frequency of nephrotic syndrome and the highest frequency of hypertension. Although these patients had good renal survival, their complete and partial remission was poor. This lesion did not have a predilection for African Americans. Thus, Fig. 2.1c is consistent with option 3 (perihilar FSGS).
The clinical features of patients with FSGS NOS are similar to those of the perihilar FSGS. Hypertension and nephrotic syndrome were common, and complete remission was low. Glomerulosclerosis and chronic tubulointerstitial injury were not severe. Therefore. Fig. 2.1d is consistent with option 4 (FSGS NOS).
The frequency of cellular FSGS was only 3 %. Therefore, no conclusions regarding clinical features or renal outcomes could be reached with this lesion (Figure not shown).
In summary, the clinical features and complete remission were worst in patients with collapsing FSGS, and more favorable in patients with tip lesion FSGS.
Suggested Reading
Thomas DB, Franceschini N, Hogan SL, et al., Clinical and pathologic characteristics of focal segmental glomerulosclerosis pathologic variants. Kidney Int 69:920–926, 2006.
D’Agati V. The spectrum of focal segmental glomerulosclerosis: new insights. Curr Opin Nephrol Hypertens 17:271–281, 2008.
Fogo AB, Kashgarian M: Diagnostic Atlas of Renal Pathology, 2nd ed, Philadelphia, Elsevier/Saunders, 2012, pp. 28–41.
10.
According to the Oxford classification of IgA nephropathy , which one of the following pathologic features is LEAST predictive of the rate of eGFR decline and ESRD?
A.
Mesangial hypercellularity score
B.
Segmental sclerosis or adhesion, interstitial fibrosis/tubular atrophy
C.
Endocapillary hypercellularity
D.
Cellular and fibrocellular crescents
E.
C and D
The answer is E
Although IgA nephropathy is the most common primary glomerular disease worldwide, there is no international consensus for its pathologic or clinical classification. Recently, a new classification, called the Oxford classification of IgA nephropathy , was introduced to identify specific pathologic features that would predict the risk for progression of the disease and treatment response to ACE-Is and/or ARBs as well as immunosuppressive agents. This new Oxford classification of IgA nephropathy is based on 265 renal biopsy sections with clinical data that were collected internationally from Asia, Europe, North America and South America. Initially, seven histopathological features were identified for reproducibility:
1.
Mesangial cell hypercellularity (>4 mesangial cells)
2.
Segmental sclerosis or adhesion
3.
Global glomerulosclerosis
4.
Endocapillary hypercellularity (hypercellularity due to increased number of cells within glomerular capillary lumina, causing narrowing of the lumina)
5.
Cellular or fibrocellular crescents
6.
Interstitial fibrosis/tubular atrophy
7.
Arteriosclerosis
However, only four independent histopathological features with reproducibility and predictive power were selected:
1.
Mesangial hypercellularity
2.
Segmental sclerosis or adhesion
3.
Endocapillary hypercellularity
4.
Interstitial fibrosis/tubular atrophy
The association between the above histopathological features and renal prognosis was retrospectively assessed during 69 months of median follow-up period. The clinical endpoints were a 50 % decline in eGFR and the development of ESRD. During the 69 months follow-up period, 22 % of them developed a 50 % reduction in eGFR and 13 % reached ESRD. By multivariate linear regression model, only segmental sclerosis or adhesion and interstitial fibrosis/tubular atrophy but not mesangial cell hypercellularity were strongly associated with decline in eGFR after adjustment for baseline or follow-up mean arterial pressure, proteinuria and eGFR . By Cox proportional hazard models, mesangial hypercellularity and interstitial fibrosis/tubular atrophy but not segmental sclerosis or adhesion were associated with a decline in eGFR and ESRD. Considering both models, only mesangial cell hypercellularity, segmental sclerosis or adhesion and interstitial nephritis/tubular atrophy have significant predictive power of renal prognosis.
Univariate models have suggested that endocapillary hypercellularity is likely to be associated with responsiveness to treatment with immunosuppressive agents. Also, fibrocellular crescents have no prognostic importance. Thus, option E is correct.
Based on the above results, the Oxford classification recommends reporting mesangial cell hypercellularity, endocapillary proliferation, segmental sclerosis or adhesion, and interstitial fibrosis/tubular atrophy as histopathological prognostic features in a biopsy specimen of a patient with IgA nephropathy.
Suggested Reading
Cattran DC, Coppo R, Cook HT, et al. The Oxford classification of IgA nephropathy: rationale, clinicopathological correlations, and classification. Kidney Int 76:534–545, 2009.
Roberts ISD, Cook HT, Troyanov S, et al. The Oxford classification of IgA nephropathy: pathology, definitions, correlations, and reproducibility. Kidney Int 76:546–556, 2009.
11.
Match the most appropriate initial treatment of the following renal diseases with proteinuria >3.5 g/day:
A.
Minimal change disease in children
B.
Minimal change disease in adults
C.
Idiopathic focal segmental sclerosis (FSGS) other than collapsing variant
D.
Idiopathic membranous nephropathy with proteinuria <2 g/day
1.
Steroids for >8 weeks
2.
Steroids for 8 weeks
3.
Steroids initially, then cyclosporine for steroid-resistant patients
4.
A recent meta-analysis found no benefit of steroids either on remission of proteinuria or preservation of renal function
Answers: A = 2; B = 1; C = 3; D = 4 or (1 = B; 2 = A; 3 = C; 4 = D; 5 = E)
The most common cause of idiopathic nephrotic syndrome in children is minimal change disease, which responds to corticosteroids . Patients are treated initially with prednisone without a renal biopsy. The protocols that include optimal dose, duration of treatment, and the route of administration are variable. However, one approach is to start prednisone at 60 mg/m 2 /day (maximum 80 mg/day) for 4 weeks, and follow the proteinuric response. If there is a response, the dose should be reduced to 35–40 mg/m 2 /day on alternate days for 4–8 weeks, and then taper off over 4–6 weeks. If there is a relapse, prednisone should be started again as above. If there are frequent relapses, a renal biopsy is required. If the biopsy confirms minimal change disease, either cyclosporine (4–6 mg/kg/day) for 1 year, or cyclophosphamide (2 mg/kg/day) for 8–12 weeks should be considered.
If proteinuria does not respond after initial 4 weeks of treatment, and the renal biopsy shows minimal change disease, either cyclosporine or cyclophosphamide at the above doses should be started.
Adult patients over the age of 50 years with minimal change disease require >8 weeks of prednisone therapy. The relapse rate is much less in adults following complete remission by steroid therapy.
The degree of proteinuria determines the prognosis of renal survival in patients with idiopathic FSGS. It has been shown that about 50 % of patients with proteinuria <10 g/day progress to ESRD in 5–10 years; however, patients with proteinuria >10 g/day may progress to ESRD within 5 years. Thus, patients with idiopathic FSGS may benefit from reduction in proteinuria. One approach is to start prednisone at 1 mg/kg/day (maximum 80 mg/day) or 2 mg/kg on alternate days for 3–4 months in patients with nephrotic syndrome and serum creatinine level <2–3 mg/dL. If there is a response, prednisone should be tapered over a period of 3–6 months. Steroid should be tried again for a relapse after prolonged remission. If there is no response after 4 weeks, cyclosporine at 2–4 mg/kg/day (~100 mg twice daily) should be tried for at least 6 months with concurrent use of prednisone (15 mg/day). Note that continuation of low-dose steroid (15 mg/day) may potentiate the response to cyclosporine. A recent study showed that the addition of chlorambucil to prednisone did not improve proteinuria or renal function in patients with idiopathic FSGS . However, this study did not report the histologic variants of FSGS.
Unlike FSGS, patients with idiopathic membranous nephropathy do not usually respond to steroids alone. A recent meta-analysis found that corticosteroids conferred no benefit in reducing proteinuria or preserving renal function. On the other hand, a beneficial effect of alkylating agents was found with cyclophosphamide giving fewer adverse reactions than chlorambucil. The recommended regimen is oral prednisone (0.5 mg/kg/day) or methylprednisolone I g IV for 3 days only on months 1, 3, and 5 and oral cyclophosphamide (2–2.5 mg/day) on months 2, 4, and 6. If cyclosporine or tacrolimus is preferred, cyclosporine should be started at 3–5 mg/kg/day in 2 divided doses for at least 6 months. Cyclosporine levels should be monitored and maintained its trough levels between 120 and 200 μg/L. Concomitant use of prednisone at 10 mg every other day is found to be useful along with cyclosporine. Also, synthetic ACTH seems to have a beneficial effect on proteinuria and renal function in patients with membranous nephropathy.
Suggested Reading
Dember LM, Salant DJ: Minimal change disease. In Wilcox CS (ed): Therapy in Nephrology and Hypertension, Philadelphia, Saunders/Elsevier, 2008, pp. 205–219.
Cho ME, Kopp JB: Focal segmental glomerulosclerosis and collapsing glomerulopathy. In Wilcox CS (ed): Therapy in Nephrology and Hypertension, Philadelphia, Saunders/Elsevier, 2008, pp. 220–238.
D’Agati V D, Kaskel FJ, Falk RJ. Focal segmental glomerulosclerosis. N Engl J Med 365:2398–2411, 2011.
Nachman PH, Jennette JC, Falk RJ. Primary glomerular diseases. In Taal MW, Chertow GM, Marsden PA, et al. (eds): Brenner & Rector’s The Kidney, 9th ed, Philadelphia, Elsevier Saunders, 2012, pp. 1100–1191.
Schnaper HW, Kopp JB. Nephrotic syndrome and the podocytopathies: Minimal change nephropathy, focal segmental glomerulosclerosis, and collapsing glomerulopathy. In Coffman TM, Falk RJ, Molitoris BA, et al. (eds) Schrier’s Diseases of the kidney 9th ed, Philadelphia, Wolters Kluwer/Lippincott Williams & Wilkins, 2013, pp. 1414–1521.
12.
Rituximab , a monoclonal antibody directed against CD20+, has been found to be LEAST likely useful in the treatment of which one of the following renal diseases?
A.
Idiopathic membranous nephropathy
B.
Minimal change disease
C.
Class IV lupus nephritis
D.
ANCA-positive renal disease
E.
Diabetic nephropathy
The answer is E
Rituximab, a monoclonal antibody directed against CD20+ antigen, has been successfully used as a therapeutic agent in a variety of renal diseases, including idiopathic membranous nephropathy, cryoglobulinemia-associated MPGN, class IV lupus nephritis , and ANCA- positive renal diseases . The CD20+ antigen is found on immature and mature B cells, as well as on malignant B cells, and rituximab treatment has been shown to prevent B cells from proliferation because of apoptosis and lysis through complement-dependent and complement-independent mechanisms. Rituximab might also inhibit T-cell activation. Thus, rituximab affects production of antibodies as well as regulation of immunoglobulin maturation by B cells. Improvement in proteinuria, and vaculitis, as well as disappearance of HCV has been observed with rituximab therapy. Rituximab has also been used in renal transplant patients to lower alloreactive antibodies, and to treat rejection associated with B cells and antibodies. Steroid-dependent nephrotic syndrome due to minimal change disease is found to respond to rituximab. However, rituximab has not been used in patients with diabetic nephropathy. Thus, choice E is incorrect. It should be noted that rituximab can cause fatal pulmonary fibrosis.
Suggested Reading
Gilbert RD, Hulse E, Rigden S. Rituximab therapy for steroid-dependent minimal change nephrotic syndrome. Pediatr Nephrol 21:1698–1700, 2006.
Salama AD, Pusey CD: Drug insight: rituximab in renal disease and transplantation. Nature Clin Pract Nephrol 2:221–230, 2006.
Francois H, Daugas E, Bensman A, et al. Unexpected efficacy of rituximab in multirelapsing minimal change nephrotic syndrome in the adult: First case report and pathophysiological considerations. Am J Kid Dis 49:158–161, 2007.
Ahmed MS, Wong CF. Rituximab and nephrotic syndrome: a new therapeutic hope? Nephrol Dial Transplant 23:11–17, 2008.
Jones RB, Tervaert JWC, Hauser T, et al. Retuximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med 363:211–220, 2010.
12a.
A 16-year-old male student is diagnosed with Alport syndrome. Which one of the following statements is FALSE regarding this syndrome?
A.
Antisera or monoclonal antibodies to type-IV collagen chains reveal loss of immunoreactivity for the α5 and α3 chains from the glomerular basement membrane (GBM) of males with X-linked disease
B.
Proteinuria and hypertension increase with age and occur much more likely in affected males than females in X-linked disease
C.
X-linked male patients with deletions in α5 chain of type-IV collagen progress to ESRD during the second or third decade of life with deafness
D.
Male patients with autosomal dominant disease with heterozygous mutations in α3 or α4 subunits of type-IV collagen will have gross hematuria and rapid progression to ESRD
E.
Autosomal recessive disease has no expression or immunostaining for α3 or α4 chain of type-IV collagen in the GBM
The answer is D
Alport syndrome is a progressive hereditary disorder in children or young adults with characteristic ultrastructural changes of the glomerular basement membrane (GBM) associated with hearing loss and eye abnormalities. There are three genetic forms of Alport syndrome. About 80 % of individuals have X-linked form of the disease. Approximately 15 % have an autosomal recessive form, and about 5 % have an autosomal dominant form of Alport syndrome. All three forms of the disease are caused by mutations in type-IV collagen. Type-IV collagen is found in all GBMs, Bowman’s capsule, distal tubular basement membranes, epidermal basement membrane, lens capsule, and cochlea.
Type-IV collagen consists of six α-chains, designated α1 to α6. Each α chain is encoded by one gene. For example α1 chain, usually designated α1 (IV), is encoded by the gene COL4A1. Thus, there are six genes that are located in three different chromosomes and encode all six chains. Each α-chain has a collagenous and noncollagenous domain. Three α-chains fold at the collagen domain to form a triple helix, and only three sets of triple helical molecules can be formed from all six chains and called promoters . These promoters have the composition of 1.1.2, 3.4.5, and 5.5.6. chains. The promoters, in turn, interact to form three types of collagen networks: 1.1.2/1.1.2 in all basement membranes, 3.4.5/3.4.5 in GBMs, tubular basement membranes, eye, and cochlea, and 1.1.2/5.5.6 in skin and Bowman capsule.
X-linked Alport syndrome is due to mutations in COL4A5, the gene encoding the α5 chain of type-IV collagen . Over 200 mutations have been reported in this form of Alport syndrome. Mutations in both alleles of COL4A3, encoding the α3 chain, or COL4 A4, encoding the α4 chain of type-IV collagen, cause autosomal recessive form of Alport syndrome, whereas heterozygous mutations in the gene COL4A3 or COL4A4 are associated with autosomal dominant Alport syndrome. The association of X-linked Alport syndrome with leiomyomatosis of the esophagus or tracheobronchial tree is due to large deletions that span the adjacent 5′ ends of the COL4A5 and COL4A6 genes.
Genetic screening for Alport syndrome is extremely difficult because of several mutations that cause the disease. However, staining for various chains of type-IV collagen has become a routine procedure to establish the diagnosis of Alport syndrome. Patients with X-linked Alport syndrome have absent or negative staining for α5 chain, which is accompanied by loss of α3 chain in the kidney. In the autosomal recessive form, staining for α3 and α4 chains is absent in the kidney. Patients with autosomal dominant form of the disease may have either normal staining or loss of α3 chain.
Microscopic hematuria is the cardinal feature of Alport syndrome . Proteinuria may be absent in early phase of the disease. However, proteinuria and hypertension develop with aging much more commonly in males than females in X-linked disease. The clinical course in X-linked males is largely dependent on the type of mutation in COL4 A5 gene. Large deletions in the gene confer a 90 % probability of ESRD before the age of 30 years, with 50 % reaching ESRD by age 20. Deafness is not congenital, but occurs during adolescence or early in about 55 % of males and 45 % of females. Ocular defects (anterior lenticonus) are restricted to patients who progress to ESRD. Autosomal recessive disease is diagnosed with similar clinical manifestations as X-linked disease, and both sexes may reach ESRD before the age of 30. Autosomal dominant disease is a much milder form compared to other forms of the disease, and patients with this disease do not progress rapidly to ESRD. However, ESRD occurs after 50 years of age. Therefore, option D is incorrect.
Suggested Reading
Hudson BG, Tryggvason K, Sundaramoorthy M, Neilson EG: Alport’s syndrome, Goodpasture’s syndrome, and type IV collagen, N Engl J Med 348:2543–2556, 2003.
Kashtan CE: Familial hematuria. Pediatr Nephrol 24:1951–1958, 2009.
Savige J, Gregory M, Gross O, et al. Expert guidelines for the management of Alport syndrome and thin basement membrane nephropathy. J Am Soc Nephrol 24:364–375, 2013.
13.
A 30-year-old man with X-linked Alport syndrome (XLAS) with deafness and anterior lenticonus and a functioning renal allograft from a living-related donor 9 months ago is concerned about his long-term function and survival of transplanted kidney. His creatinine is 1.2 mg/dL. Which one of the following diseases can occur and cause acute allograft loss in this patient?
A.
FSGS
B.
Recurrence of Alport syndrome
C.
Minimal change disease
D.
Anti-GBM antibody disease
E.
Membranous nephropathy
The answer is D
Renal transplantation, besides dialysis, is the only long-term treatment modality available for patients with XLAS. Survival rate of renal allograft in XLAS patient is similar to patients with other diseases. Recurrence of Alport syndrome does not occur in the transplant because the donor GBM is normal; however, 3–5 % of patients develop de novo anti-GBM antibody disease. Usually, anti-GBM antibody disease occurs within a year of transplantation, but occurrence after several years has been described. As stated in the previous question, the antibodies are usually directed against the α-5 chain of type-IV collagen. Affected patients have high titers of these antibodies and are at high risk for crescentic GN and loss of graft function. Treatment with plasmapheresis and cyclophosphamide is of limited benefit, and retransplantation is associated with a high recurrence rate of anti-GBM antibody disease. The occurrence of FSGS, minimal change disease, and membranous nephropathy has not been reported. Thus, option D is correct.
Suggested Reading
Byrne MC, Budisavljevic MN, Fan Z, et al. Renal transplant in patients with Alport’s syndrome. Am J Kidney Dis 39:769–775, 2002.
Kashtan CE. Renal transplantation in patients with Alport syndrome. Pediatr Transplant 10:651–657, 2006.
Gumber MR, Kute EB, Gopalani KR, et al. Outcome of renal transplantation in Alport’s syndrome: a single-center experience. Transplant Proc 44:261–263, 2012.
14.
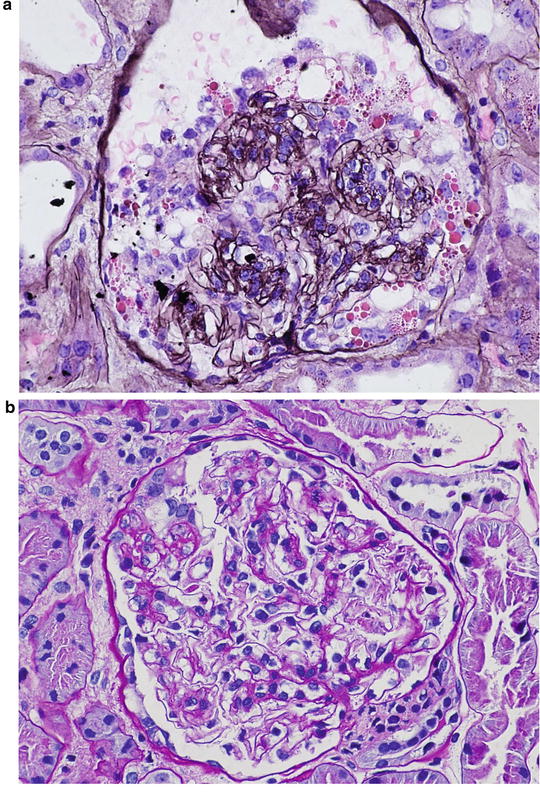
A 35-year-old Caucasian man is referred to a nephrologist for episodic hematuria. More specifically, hematuria occurs following upper respiratory tract infection. There is no history of extrarenal manifestations. Serum creatinine is normal (eGFR = 100 mL/min). Urinalysis shows microscopic hematuria and proteinuria. A spot urine protein to creatinine ratio is 0.4. A renal biopsy shows no specific lesion on LM. Also, IM studies are negative . Based on the following EM photomicrograph (Fig. 2.2 ), which one of the following is the MOST likely diagnosis?
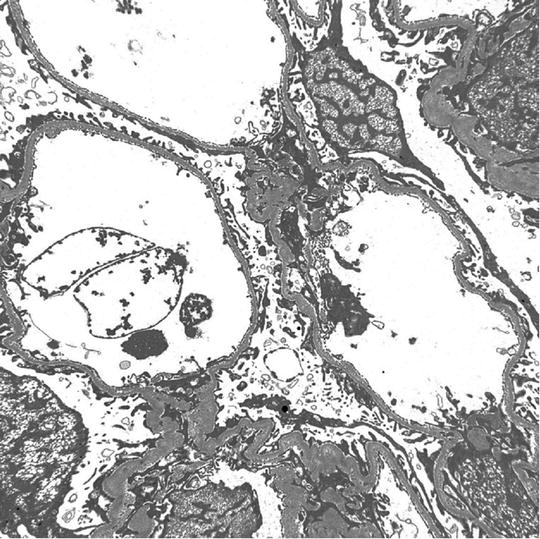
Fig. 2.2
EM micrograph of the above patient
A.
IgA nephropathy
B.
X-linked Alport syndrome
C.
Autosomal recessive Alport syndrome
D.
Thin basement membrane nephropathy
E.
Minimal change disease
The answer is D
Except for minimal change disease , all other disease conditions initially present with hematuria. The clue for diagnosis is the finding on EM, which shows a uniformly thin basement membrane. Thin basement membranes are also found in Alport syndrome and IgA nephropathy; however, these two diseases can be distinguished from thin basement membrane nephropathy by LM and IF findings and clinically. In Alport syndrome, the cardinal ultrastructural abnormality is the variable thickening, thinning, basket weaving, and lamellation of the glomerular basement membrane. According to one study, the normal range for basement membrane width in adult males is 373 ± 42 nm, and in females 326 ± 45 nm. In thin basement membrane nephropathy, the glomerular basement membrane width is <200 nm (for comparison, the normal GBM is shown on the right of the Fig. 2.3 below). At present, 40 % of thin basement membrane nephropathy is caused by mutations in COL4A3 and COL4A4 genes that encode α3 and α4 chains of type-IV collagen.
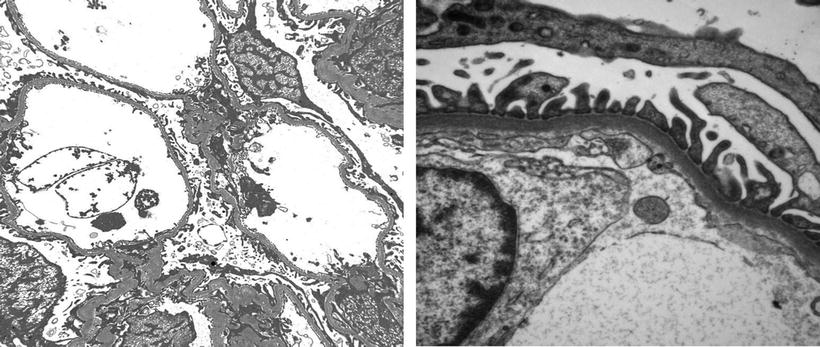
Fig. 2.3
EM micrograph showing thin basement membrane width (left) and normal basement membrane width (right)
In children, the diagnosis of thin basement membrane is rather difficult unless each laboratory establishes its own age-related normal glomerular basement membrane width. In one study, the range varied from 146 to 273 nm in a male child at 1 year of age to 230–430 nm at age 9 or older.
Suggested Reading
Savige J, Rana K, Tonna S, et al. Thin basement membrane nephropathy. Kidney Int 64:1169–1178, 2003.
Tryggvason K, Patrakka J: Thin basement membrane nephropathy. J Am Soc Nephrol 17:813–822, 2006.
Savige J, Gregory M, Gross O, et al. Expert guidelines for the management of Alport syndrome and thin basement membrane nephropathy. J Am Soc Nephrol 24:364–375, 2013.
15.
A 54-year-old Caucasian woman presents to her primary care physician for severe headache. Her BP is found to be 180/100 mmHg. Physical exam is normal except for trace edema. A urinalysis reveals hematuria, 3+ proteinuria, and RBC casts. Serum creatinine is 2.2 mg/dL and BUN 60 mg/dL. She is referred to a nephrologist. Renal biopsy (Fig. 2.4) shows:
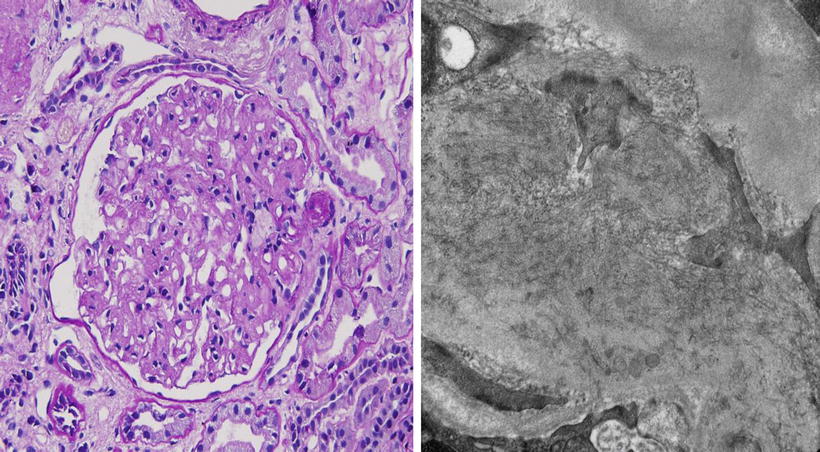
Fig. 2.4
Light (left) and electron (right) photomicrographs of the patient described above
LM (Left): mesangial cell proliferation with lobular pattern. Congo red stain-negative
EM (Right): randomly arranged fibrils ranging from 15 to 30 nm in diameter distributed throughout the glomerulus
IF: prominent, but sludgy IgG and C3 in mesangial areas with mild staining for IgM, IgA, and C1q with IgG4 dominance
Based on the above information, which one of the following is the MOST likely diagnosis?
A.
Membranoproliferative glomerulonephritis (MPGN)—type I
B.
Lupus nephritis (Class III)
C.
Monoclonal immunoglobulin deposit disease
D.
Immunotactoid glomerulopathy
E.
Fibrillary glomerulonephritis (GN)
The answer is E
Based upon the size of the fibrils, the patient has fibrillary GN rather than immunotactoid glomerulopathy. Light microscopic findings may not distinguish fibrillary GN from other glomerular diseases, as mentioned in options from A to D, because fibrillary GN is pleomorphic and present as lobular glomerulopathy as type-1 MPGN, membranous or crescentic GN. The definitive diagnosis of fibrillary GN is usually made by electron microscopy, which shows non-branching fibrils ranging from 15 to 30 nm in diameter, as opposed to immunotactoid glomerulopathy that demonstrates fibrils with >30 nm in diameter. Furthermore, immunotactoid glomerulopathy is found in association with lymphoproliferative diseases. There is no known effective therapy for fibrillary GN. Lupus nephritis and monoclonal immunoglobulin deposit disease do not demonstrate fibrils on EM .
Suggested Reading
Ronco PM, Aucouturier P, Moulin B: Renal amylopdosis and glomerular diseases with monoclonal immunoglobulin deposition. In Floege J, Johnson RJ, Feehally J (eds). Comprehensive Clinical Nephrology, 4th ed, Philadelphia, Saunders/Elsevier, 2010, pp. 322–334.
Appel GB, Radhakrishnan J, D’Agati VD: Secondary glomerular disease. In Taal MW, Chertow GM, Marsden PA, et al. (eds): Brenner & Rector’s The Kidney, 9th ed, Philadelphia, Elsevier Saunders, 2012, pp. 1192–1277.
Fogo AB, Kashgarian M: Diagnostic Atlas of Renal Pathology, 2nd ed, Philadelphia, Elsevier/Saunders, 2012, pp. 94–102.
16.
A 15-year-old female was brought to the physician by her parents for loss of fat in the upper part of the body, rapid growth (5′ 11″ tall), muscular hypertrophy, subcutaneous nodules and macroglossia. Urinalysis showed proteinuria and hematuria. A spot urine protein to creatinine ratio was 3.2. C4 was normal, but C3 was very low. Serum creatinine was 1.4 mg/dL. A renal biopsy showed the following:
LM: endocapillary proliferation with thickened glomerular basement membranes (GBM). The involved portions of the GBM resemble a “string of sausages”
EM: dense deposits in the basement membrane, Bowman capsule and tubules
IF: C3 staining irregular along the capillary wall. Immunoglobulins absent (Figs. 2.5, 2.6, and 2.7)
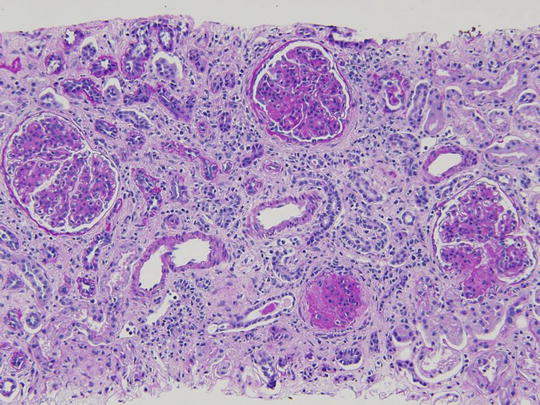
Fig. 2.5
Pattern of glomerular injury observed by light microscopy
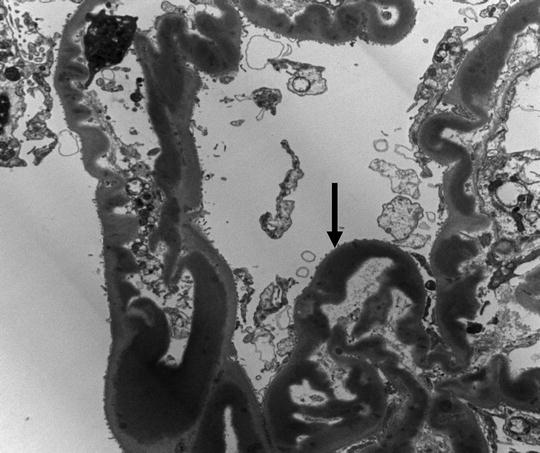
Fig. 2.6
Electron micrograph (EM) showing dense appearance of the GBM (arrow)
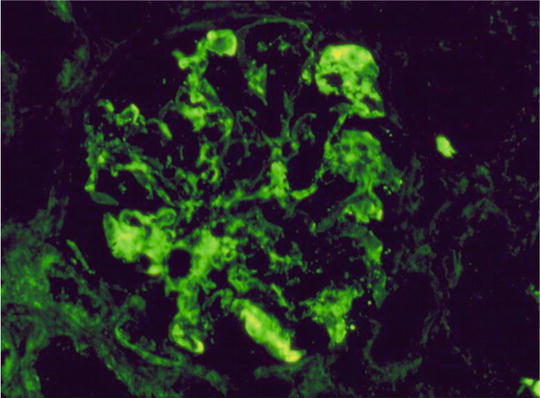
Fig. 2.7
Immunofluorescence (IF) staining pattern (irregular) of C3
The clinical presentation and renal pathology findings are MOST consistent with which one of the following diagnosis?
A.
Lupus nephritis (Class IV)
B.
Membranoproliferative glomerulonephritis (MPGN type I )
C.
Dense deposit disease (MPGN type II)
D.
Amyloidosis
E.
C1q nephropathy
The answer is C
The patient described in the question has partial lipodystrophy (PLD) , which is most commonly seen in girls between 5 and 15 years of age. The most common renal disease that is associated with PLD is MPGN II (type II or dense deposit disease). Therefore, option C is correct. Renal disease occurs in 20–50 % of patients with PLD, and PLD occurs in 10 % of patients with MPGN type II. Apart from renal disease, patients with partial lipodystrophy, similar to total lipodystrophy patients, may have several metabolic and systemic abnormalities, including tall stature, muscular hypertrophy, subcutaneous nodules, macroglossia, hyperinsulinemia, insulin resistance, and diabetes.
The renal biopsy findings with low C3 and normal C4 are consistent with the dense deposit disease (MPGN, type II). Patients are noted to have asymptomatic proteinuria and microhematuria , but nephrotic syndrome is occasionally present. The pathogenesis of the acquired form of PLD is believed to be an autoimmune disorder. It has been reported that MPGN II or PLD or both are associated with dysfunction of the complement system. Subsequently, an IgG autoantibody was detected called the C3 nephritic factor [C3NeF] . The target of this autoantibody is the alternative pathway C3 convertase C3bBb. Thus, diminished complement C3 levels in association with the C3NeF is the most prominent serologic abnormality in patients with dense deposit disease and PLD. The glomerular disease progresses rapidly to ESRD. Plasmapheresis has been shown to be beneficial in patients with C3NeF.
In MPGN I and lupus nephritis , both C4 and C3 are generally depressed, but the complement levels are normal in patients with amyloidosis and C1q nephropathy .
Suggested Reading
Smith RJH, Harris CL, Pickering MC. Dense deposit disease. Mol Immunol 48:1604–1610, 2011.
Fogo AB, Kashgarian M: Diagnostic Atlas of Renal Pathology, 2nd ed, Philadelphia, Elsevier/Saunders, 2012, pp. 53–61.
Nachman PH, Jennette JC, Falk RJ. Primary glomerular diseases. In Taal MW, Chertow GM, Marsden PA, et al. (eds): Brenner & Rector’s The Kidney, 9th ed, Philadelphia, Elsevier Saunders, 2012, pp. 1100–1191.
Sethi S, Fervenza FC. Membranoproliferative glomerulonephritis-A new look at an old entity. N Engl J Med 366:1119–1131, 2012.
17.
Match the following disease states with the electron microscopy findings of the glomeruli:
1.
Amyloidosis
2.
Immunotactoid glomerulopathy
3.
Fibrillary glomerulonephritis (GN)
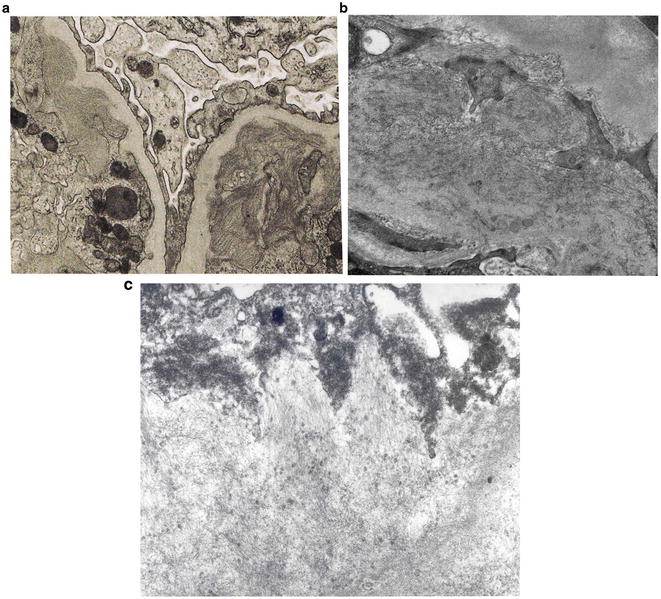
Light microscopy does not distinguish amyloidosis from either fibrillary or immunotactoid GN . All three diseases will give the appearance of nodules or lobules resembling diabetic glomerulosclerosis or membranoproliferative GN. However, these diseases can be distinguished by the size of the fibrils seen on electron microscopy. The fibrils in amyloidosis are randomly arranged and range from 8 to 12 nm in diameter (Fig. 2.8c ), whereas in fibrillary or immunotactoid GN the diameters of the fibrils range from 15 to 30 nm (Fig. 2.8b ) and 30–50 nm (Fig. 2.8a ), respectively. In fibrillary GN, the fibrils are also randomly arranged as in amyloidosis, but they are larger. In immunotactoid GN, the fibrils appear as large microtubular deposits and are arranged in parallel arrays giving the appearance of a “stacked wood” arrangement (Fig. 2.8a ).
Suggested Reading
Ronco PM, Aucouturier P, Moulin B: Renal amylopdosis and glomerular diseases with monoclonal immunoglobulin deposition. In Floege J, Johnson RJ, Feehally J (eds). Comprehensive Clinical Nephrology, 4th ed, Philadelphia, Saunders/Elsevier, 2010, pp. 322–334.
Appel GB, Radhakrishnan J, D’Agati VD: Secondary glomerular disease. In Taal MW, Chertow GM, Marsden PA, et al. (eds): Brenner & Rector’s The Kidney, 9th ed, Philadelphia, Elsevier Saunders, 2012, pp. 1192–1277.
Fogo AB, Kashgarian M: Diagnostic Atlas of Renal Pathology, 2nd ed, Philadelphia, Elsevier/Saunders, 2012, pp. 283–286.
18.
Match the following mutations in the podocyte/slit-diaphragm protein genes with clinical findings:
A.
NPHS 1 (nephrin)
B.
NPHS 2 (podocin)
C.
ACTN4 (α-actinin-4)
D.
CD2-Associated Protein (CD2AP)
1.
Infants exhibit massive proteinuria, ascites, anasarca, polycythemia, and failure to thrive with recurrent infections
2.
An autosomal dominant form of FSGS with subnephrotic proteinuria and progressive renal insufficiency
3.
Early childhood onset of proteinuria, FSGS, and rapid progression to ESRD. Usually steroid-resistant, and the disease does not recur in transplanted kidney
4.
Nephrotic syndrome in mice and FSGS in humans
Answers: A = 1; B = 3; C = 2; D = 4
In the last 10–15 years, considerable progress has been made in our understanding of the molecular biology of podocytes and slit-diaphragms. Several proteins both in cytoskeleton of podocytes and slit diaphragms have been identified. Nephrin, podocin, CD2AP are localized to the slit-diaphragms, whereas α-actinin-4 is present in the cytoskeleton of the podocytes. Mutations in the nephrin gene ( NPHSI ) leads to congenital nephrotic syndrome of the Finnish types which is characterized by autosomal recessive inheritance. Infants are born with nephrotic syndrome ; if untreated, these infants develop complications of nephrotic syndrome such as infections and thrombosis. Treatment includes aggressive supportive care until nephrectomy and renal transplantation is performed.
Another protein associated with nephrin is podocin. The gene for this protein is NPHS2 . Defects in podocin cause FSGS and nephrotic syndrome , which is steroid-resistant and present in childhood, adolescence or adulthood. The disease is inherited as an autosomal recessive with progression to ESRD. Recurrence of disease in renal allograft is rather rare compared to idiopathic FSGS.
CD2AP interacts with nephrin and functions as an adaptor molecule between nephrin and actin filaments of the cytoskeleton. Mutation in the CD2AP gene causes nephrotic syndrome in mice. Also, splice mutations of the gene have been identified in patients with FSGS,
Mutations in the gene ACTN4 which encodes α-actinin-4 causes an autosomal dominant form of FSGS with subnephrotic proteinuria and gradual progression to chronic kidney disease. It rarely recurs in the renal allograft.
The following table summarizes the characteristics of the above discussed genes and their proteins (Table 2.1 ).
Table 2.1
Gene mutation and nephrotic syndrome
Gene | Inheritance | Protein | Age of subject | Disease and their features |
|---|---|---|---|---|
NPHS1 | Autosomal recessive | Nephrin | Infancy | Congenital nephrotic syndrome of the Finnish type |
NPHS2 | Autosomal recessive | Podocin | 3 Months to adulthood | Steroid-resistant nephrotic syndrome/FSGS |
CD2AP | Autosomal dominant | CD2AP | Adult | FSGS/nephrotic syndrome |
ACTN4 | Autosomal dominant | α-Actinin-4 | Adult | Subnephrotic proteinuria, nephrotic syndrome/FSGS |
Suggested Reading
Mundel P, Shankland SJ. Podocyte biology and response to injury. J Am Soc Nephrol 13:3005–3015, 2002.
Pavenstädt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev 83:253–307, 2003.
Johnstone DB, Holzman LB. Clinical impact of research on the podocyte slit diaphragm. Nature Clin Pract Nephl 2:271–282, 2006.
Tryggvason K, Patrakk J, Wartiovaara J. Hereditary proteinuria syndromes and mechanisms of proteinuria. N Engl J Med 354:1387–1401, 2006.
19.
Match the following LM pictures with the clinical scenarios:
A.
A 56-year-old Caucasian man with isolated nephrotic syndrome
B.
A 10-year-old boy with nephrotic syndrome
C.
An 18-year-old woman with hematuria and proteinuria and low C3
D.
A 19-year-old African American man with nephrotic syndrome
Answers: A = Fig. 2.9d & e; B = Fig. 2.9a ; C = Fig. 2.9c ; D = Fig. 2.9b
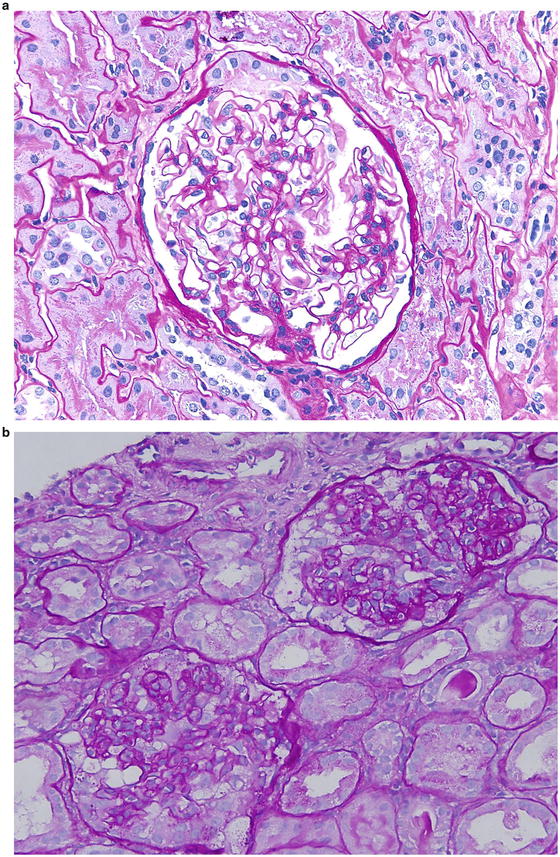
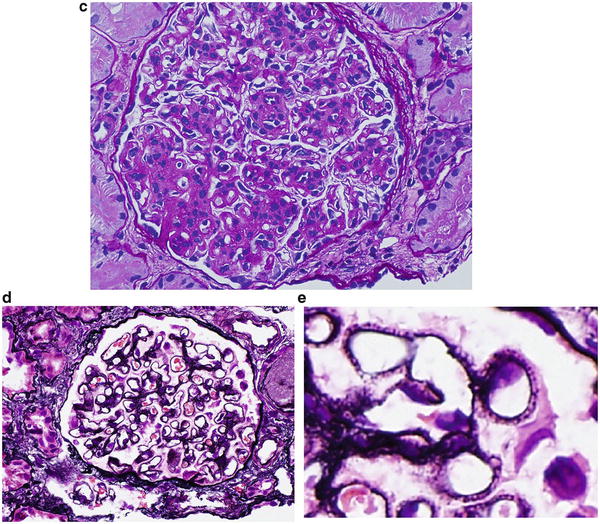
Fig. 2.9
Pattern of glomerular injury observed by light microscopy
The frequent cause of isolated nephrotic syndrome in a 56-year-old Caucasian man is membranous nephropathy. The “spike and dome” appearance of the GBM shown in Fig. 2.9d , e are consistent with the presentation of the patient.
The most common cause of nephrotic syndrome in children is minimal change disease , and the glomerulus appears normal on light microscopy. Figure 2.9a shows normal glomerular appearance, which is consistent with the description of the patient in choice B.
Figure 2.9c shows hypercellularity, mostly mesangial cells and infiltrating leukocytes and lobular appearance, consistent with MPGN type 1. Patients often have low C3 and total hemolytic complement (CH50). Thus, the description of Fig. 2.9c is consistent with the presentation of the patient given in choice C.
The most likely glomerular disease in the 19-year-old African American male with nephrotic syndrome is collapsing variant of FSGS, and Fig. 2.9b is consistent with this diagnosis.
Suggested Reading
Fogo AB, Kashgarian M: Diagnostic Atlas of Renal Pathology, 2nd ed, Philadelphia, Elsevier/Saunders, 2012, pp. 1–550.
Nachman PH, Jennette JC, Falk RJ. Primary glomerular diseases. In Taal MW, Chertow GM, Marsden PA, et al. (eds): Brenner & Rector’s The Kidney, 9th ed, Philadelphia, Elsevier Saunders, 2012, pp. 1100–1191.
20.
A 22-year-old African American woman with HIV disease presents to the Emergency Department with nausea, vomiting, hematuria, proteinuria, and swollen legs. She used heroin and cocaine 7 years ago. She got HIV transmission from a sexual contact. Pertinent labs include a creatinine level of 3.4 mg/dL, and albumin concentration of 2.4 g/dL. ANA was weakly positive (titer <1:80). Anti-double-stranded DNA is negative. Serum complements are normal. A 24 h urine protein reveals 6.5 g. The kidneys are large on renal ultrasound. A renal biopsy shows:
LM: diffuse glomerulonephritis with endocapillary cell proliferation (Class IV)
EM: mesangial and subendothelial electron-dense deposits with tubuloreticular inclusion bodies in endothelial cells
IF: granular glomerular staining (>1+) for IgG, IgM, IgA, C3, C1q, κ and λ chains
The biopsy results are consistent with which one of the following glomerular diseases?
A.
Focal segmental glomerulosclerosis (FSGS) of collapsing variant
B.
IgA nephropathy
C.
Lupus-like glomerulonephritis (GN)
D.
Immune complex GN other than lupus-like GN
E.
Minimal change disease
The answer is C
Classically, FSGS of collapsing variant is the most common glomerular lesion found in HIV patients. IF staining is usually absent. This classic lesion is generally referred to as HIV-associated glomerulopathy (HIVAN). Other nonimmunologic disease associated with HIV nephropathy is minimal change disease.
In addition to HIVAN, other common glomerular lesions with immune complexes have been described in HIV patients. These glomerular lesions fall into three major categories as HIV immune complex diseases: (1) diffuse proliferative GN or lupus-like disease; (2) IgA nephropathy; and (3) membranous nephropathy. The common histologic features of these three glomerular diseases are the presence of mesangial and glomerular basement membrane immune complexes associated with HIV antigens.
The findings described above are consistent with lupus-like GN rather than other glomerular diseases. Haas et al. reported 14 HIV patients with lupus-like GN (Class IV), characterized by a “full house” pattern of immunoglobulin and complement deposition in glomeruli. However, these patients had no strong serology for lupus. However, some patients had weakly positive (titer <1:80) ANA titers, but no anti-double-stranded DNA. Thus, lupus-like GN with immunohistologic features but absent serologic markers of lupus is another glomerular disease associated with HIV/AIDS patients.
Most of these patients have long-standing (>10 years) HIV viremia. Clinical features include hematuria, proteinuria, hypertension, and decreased renal function. Renal outcome is poor in these patients.
Suggested Reading
Haas M, Kaul S, Eustace JA: HIV-associated immune complex glomerulonephritis with “lupus-like” features. A clinicopathologic study of 14 cases. Kidney Int 67:1381–1390, 2005.
Cohen AH, Nast CC: Renal injury associated with human immunodeficiency virus infection. In Jennette JC, Olson JL, Schwartz MM, Silva FG (eds): Heptinstall’s Pathology of the Kidney. 6th ed. Philadelphia, Lippincott Williams & Wilkins, 2006, pp. 397–422.
21.
Regarding the treatment of HIVAN, which one of the following statements is INCORRECT?
A.
Antiretroviral therapy is effective in improving renal function and proteinuria
B.
Antiretroviral therapy reduces the rate of progression of kidney disease to ESRD by a significant percentage
C.
Improvement in renal function with antiretroviral therapy correlates with the reversal of histologic changes of HIVAN
D.
Antiretroviral therapy continues to have beneficial effects on renal function even after its discontinuation
E.
ACE-Is and ARBs reduce proteinuria and improve renal survival
The answer is D
The treatment and prevention of HIVAN is based on the premise that the glomerular lesions are caused by active viral infection of the kidney. Therefore, antiretroviral therapy is considered the major therapeutic modality for HIVAN. It was shown that treatment with antiretroviral agents immediately after diagnosis of HIVAN is effective in improving renal function and proteinuria, and reducing the progression of kidney disease to ESRD by 38 %. It has also been shown that the observed improvement in GFR by antiretroviral agents correlates with an improvement in histologic changes of the kidney. Loss of podocyte markers such as synaptopodin was reacquired following antiretroviral therapy. Conservative management of proteinuria with inhibition of renin-angiotensin system has been found to be effective in reducing proteinuria and prolonging renal survival in patients with HIVAN. Thus, ACE-Is and ARBs are found to be add-on drugs for the treatment of HIVAN.
Once antiretroviral therapy is discontinued, renal lesions progress at an accelerated rate. Therefore, continuation of antiretroviral therapy is indicated to improve both renal function and proteinuria in HIVAN . Thus, option D is incorrect.
Suggested Reading
Kalayjian RC. The treatment of HIV-associated nephropathy. Adv Chronic Kidney Dis 17:59–71, 2010.
Kupin W. Viral glomerulonephritis. In Coffman TM, Falk RJ, Molitoris BA, et al. (eds) Schrier’s Diseases of the kidney 9th ed, Philadelphia, Wolters Kluwer/Lippincott Williams & Wilkins, 2013, pp. 1292–1324.
22.
Match the following chemotherapeutic drugs with the reported renal toxicity:
A.
Semustine
B.
Cisplatin
C.
Mitomycin C
D.
Cyclophosphamide
E.
Interleukin-2
F.
Adriamycin
1.
Acute kidney injury (AKI) and tubulointerstitial disease (TID)
2.
Hemolytic uremic syndrome (HUS)
3.
Chronic kidney disease (CKD) 3 years after therapy
4.
AKI with high dose
5.
Hyponatremia and hemorrhagic cystitis
6.
Collapsing glomerulopathy
Answers: A = 3; B = 1; C = 2; D = 5; E = 4; F = 6
Semustine is a nitrosourea compound that crosses the blood–brain barrier easily, and therefore is used in malignant brain tumors. High-dose (1500 mg/m 2 ) therapy in children has been shown to cause CKD 3–5 years after completion of the treatment.
Cisplatin is an effective chemotherapeutic agent that has been used to treat many tumors, including bladder cancer. Nephrotoxicity is a troublesome complication of cisplatin, which is dose-dependent. TID with heavy proteinuria are commonly observed with hyaline droplets in the proximal tubular cells, tubular necrosis and degeneration of tubular BM. Tubular defect results in magnesium and phosphate wasting and AKI . Concomitant use of other nephrotoxins can potentiate cisplatin nephrotoxicity. Hydration with brisk diuresis and administration of sodium thiosulfate have been shown to reduce cisplatin-induced nephrotoxicity.
Mitomycin C is used in combination with 5-fluorouracil in treatment of gastrointestinal carcinoma. High doses of mitomycin C (>60 mg/m 2 ) have been shown to cause HUS.
Cyclophosphamide is active against lymphomas and hematologic malignancies . It causes hyponatremia due to its antidiuretic effect in the distal nephron without causing an increase in ADH levels. Also, cyclophosphamide causes hemorrhagic cystitis.
Interleukin-2, which has a killer cell function, causes AKI in high doses. It reduces GFR by renal vasoconstriction. Adriamycin use is associated with the development of proteinuria and also collapsing glomerulopathy.
Suggested Reading
Palmer BF, Henrich WL. Toxic nephropathy. In Brenner BM (ed): Brenner & Rector’s The Kidney, 7th ed, Philadelphia, Saunders, 2004, pp. 1625–1658.
Safirstein RL. Renal diseases induced by antineoplastic agents. In Schrier (ed). Diseases of the kidney & Urinary Tract. 8th ed, Philadelphia, Lippincott Williams & Wilkins, 2007, pp. 1068–1081.
23.
Match the following drugs with the reported nephrotoxicity:
A.
Acyclovir
B.
Foscarnet
C.
Indinavir
D.
Adefovir
E.
Pentamidine
1.
Hypocalcemia, hypomagnesemia, and hyperkalemia with prolonged therapy
2.
Hypocalcemia, hyperphosphatemia, and increased serum PTH levels
3.
AKI due to intratubular precipitation of needle-shaped crystals
4.
Crystalluria and nephrolithiasis
5.
Proximal tubular injury, low-grade proteinuria and an increase in serum creatinine level
Answers: A = 3; B = 2; C = 4; D = 5; E = 1
Acyclovir is an antiviral agent, which is excreted by the kidney. It causes AKI by intratubular precipitation of acyclovir crystals. Urinalysis shows needle-shaped crystals under polarizing light. Volume depletion precipitates ARF.
Foscarnet is used to treat CMV infection in transplant patients, and is excreted by the kidney. It causes AKI, hypocalcemia, hyperphosphatemia, and elevated PTH levels. Hydration with normal saline reduces nephrotoxicity.
Indinavir is a protease inhibitor that causes crystalluria and nephrolithiasis due to precipitation of the drugs in renal tubules. Long-term use of the drug includes interstitial inflammation, granuloma formation, interstitial fibrosis, and finally renal failure.
Adefovir is a nucleoside inhibitor that causes proximal tubular injury, resulting in phosphaturia, proteinuria, and AKI due to mitochondrial toxicity . It is excreted by the kidney.
Pentamidine is clinically used to treat Pneumocystis carinii pneumonia. Frequent doses accumulate in the kidney and causes tubular injury. Hypocalcemia, hypomagnesemia, and hyperkalemia have been reported with prolonged therapy.
Suggested Reading
Palmer BF, Henrich WL. Toxic nephropathy. In Brenner BM (ed): Brenner & Rector’s The Kidney, 7th ed, Philadelphia, Saunders, 2004, pp. 1625–1658.
Izzedine H, Launay-Vacher V, Deray D. Antiviral drug-induced nephrotoxicity. Am J Kidney Dis 45:804–817, 2005.
24.
Match the following clinical histories with the LM figures of the kidney:
A.
A 65-year-old man with nephrotic syndrome, hematuria, and hypertension. Glomerulus with multiple nodules, and these nodules are PAS-positive, but silver stain-negative. Hypocomplementemic and hepatitis C-positive
B.
A 70-year-old man with hematologic malignancy and deposits with microtubular fibrils >30 nm in diameter on EM
C.
A 22-year-old woman with hematuria, hypertension and low complement levels, 2 weeks following an upper respiratory tract infection
D.
A 20-year-old woman with hematuria, proteinuria, and normal complement levels, 2 days following an upper respiratory infection
Answers: A = Fig. 2.10c ; B = Fig. 2.10b ; C = Fig. 2.10d ; D = Fig. 2.10a
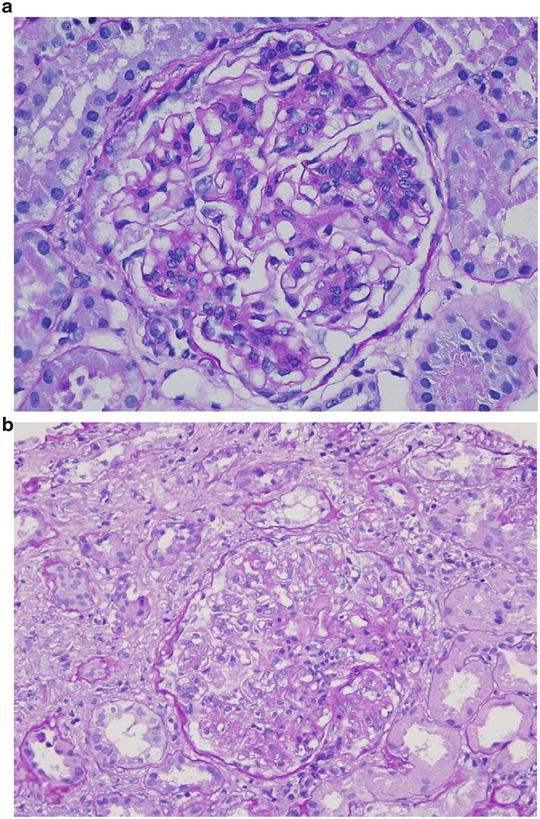
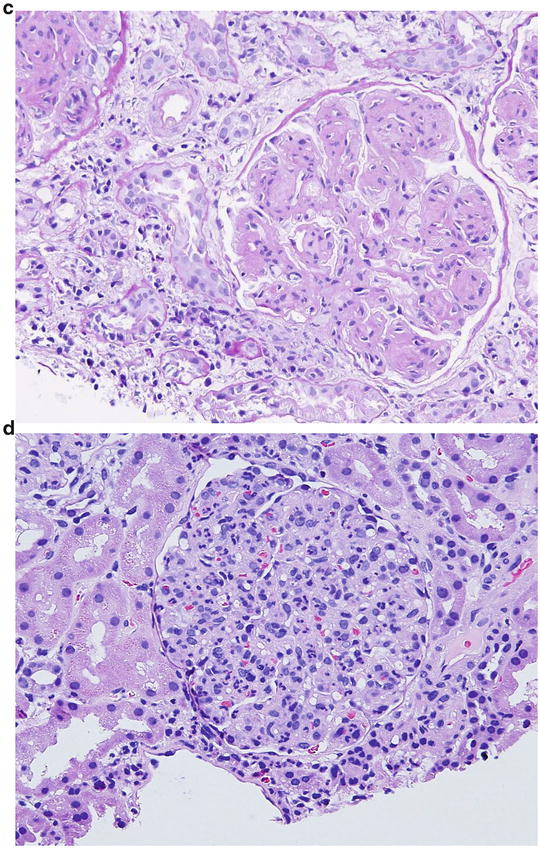
Fig. 2.10
Pattern of glomerular injury observed by light microscopy
The diagnosis in the 69 year-old man (choice A) with nephrotic syndrome, hematuria, hypertension, hypocomplementemia, and hepatitis C with glomerular nodules on renal biopsy seems to have monoclonal immunoglobulin deposition disease , which comprises light, heavy, or light-and-heavy chains. Although the clinical and pathologic characteristics are similar in all three diseases, hypocomplementemia and false-positive hepatitis C antibody is characteristic of heavy chain deposition disease ( HCDD) . Thus, Fig. 2.10c is consistent with HCDD.
The patient described in choice B seems to have immunotactoid glomerulopathy. Renal biopsy in patients with immunotactoid glomerulopathy shows either membranous nephropathy or MPGN type 1 . Thus, light microscopy of the glomerulus shows lobular pattern with nodule-like appearance in some patients. Generally, the nodules are silver-stain-negative. Figure 2.10b is consistent with immunotactoid glomerulopathy.
The clinical features of the patient in choice C are consistent with acute post-streptococcal glomerulonephritis, and the glomerular findings shown in Fig. 2.10d are consistent with this diagnosis. The glomerular lesions are characterized by diffuse endocapillary (mesangial and endothelial) hypercellularity and infiltration of leukocytes. IF microscopy shows the deposition of C3, IgG, IgM, and properdin in glomerular capillary loops and mesangium.
Figure 2.10a , by process of elimination, is compatible with the description of the patient given in choice D. This patient carries the diagnosis of IgA nephropathy. The earliest light microscopy expression of IgA nephropathy is either focal or diffuse mesangial and in some cases endothelial hypercellularity. Figure 2.10a shows segmental mesangial cell hypercellularity with deposition of mesangial matrix. Influx of leukocytes is also seen in some patients. The glomerular lesions are similar to those seen in class II lupus nephritis .
Suggested Reading
Fogo AB, Kashgarian M: Diagnostic Atlas of Renal Pathology, 2nd ed, Philadelphia, Elsevier/Saunders, 2012, pp. 1–550.
Nachman PH, Jennette JC, Falk RJ. Primary glomerular diseases. In Taal MW, Chertow GM, Marsden PA, et al. (eds): Brenner & Rector’s The Kidney, 9th ed, Philadelphia, Elsevier Saunders, 2012, pp. 1100–1191.
25.
An 82-year-old obese Caucasian woman with history of hypertension and proteinuria of 1.5 g/day. She is on ACE-I and a β-blocker. Her glycated HgbA1c is 6 %, and not on any oral hypoglycemic medications. You are asked to see the patient for an increase in proteinuria to 4.6 g/day. Urinalysis is positive for albumin without RBCs or WBCs. Which one of the following diagnoses is LEAST likely on a renal biopsy ?
A.
Membranous nephropathy
B.
Amyloidosis
C.
Minimal change disease
D.
Focal segmental glomerulosclerosis (FSGS)
E.
Fibrillary glomerulonephritis (GN)
The answer is E
Studies on renal biopsy in the very elderly (≥80 years) are sparce. Renal abnormalities in these elderly have been attributed to aging, hypertension, and other conditions. Nair et al. identified, in a retrospective analysis, 100 patients aged 80 years or older with renal biopsies for various reasons. Crescentic GN was the most common glomerular lesion, and membranous nephropathy accounted for only 15 %. Of the 100 biopsies, 40 of them showed a renal condition that would benefit from therapeutic intervention, and the remaining biopsies provided useful information that can prevent harmful intervention. Thus, a renal biopsy in the very elderly is needed for the diagnostic, prognostic, and therapeutic purpose.
Moutzouris et al. performed an analysis of another retrospective renal biopsy study in 235 elderly (≥80 year) patients and the results were compared to a control group of 264 patients aged between 60 and 61 years. The indications for biopsy were AKI, chronic progressive kidney disease, nephrotic syndrome, nephrotic syndrome with AKI , and isolated proteinuria. The most common diagnoses are shown in the following Table (Table 2.2 ).
Table 2.2
Renal biopsy findings in the very elderly with various indications for biopsy
Disease | Percent |
|---|---|
Pauci-immune GN | 19 |
Hypertensive nephrosclerosis | 7.1 |
FSGS secondary to hypertension & aging | 7.6 |
IgA nephropathy | 7.1 |
Membranous nephropathy | 7.1 |
Amyloidosis | 5 |
Minimal change disease | 5 |
Myeloma cast nephropathy | 5 |
Interestingly, renal biopsies done in patients with nephrotic syndrome alone had different diagnoses (Table 2.3 ).
Table 2.3
Renal biopsy findings in the very elderly with nephrotic syndrome
Disease | Percent |
|---|---|
Membranous nephropathy | 22 |
Amyloidosis | 18 |
Minimal change disease | 16 |
IgA nephropathy | 6 |
Pauci-immune GN | 4 |
Membranoproliferative GN | 4 |
Diabetic glomerulosclerosis | 4 |
FSGS (primary) | 4 |
Overall, the renal biopsy study in the very elderly provided diagnostic information that modified the treatment in 67 % of those patients with AKI and nephrotic syndrome. Thus, renal biopsy should be done in the very elderly, if indicated.
In the study of Uezono et al., the elderly (65 years and older) had FSGS (23 %), minimal change disease (19 %), and membranous nephropathy (15 %) as the primary diagnosis for their nephrotic syndrome. Also, 71 % of the elderly who presented with AKI had MPO-ANCA-positive crescentic GN on renal biopsy.
Fibrillary GN in the very elderly is unlikely, because the median age is around 50 years. Thus, option E is the correct answer.
Suggested Reading
Nair R, Bell JM, Walker PD. Renal biopsy in patients aged 80 years and older. Am J Kidney Dis 44:618–626, 2004.
Uezono S, Hara S, Sato Y, et al. Renal biopsy in elderly patients: A clinicopathological analysis. Ren Fail 28:549–555, 2006.
Moutzouris D-A, Herlitz L, Appel GB, et al. Renal biopsy in the very elderly. Clin J Am Soc Nephrol 4:1073–1082, 2009.
26.
Idiopathic membranous nephropathy is now considered an antibody (Ab)-mediated autoimmune glomerular disease with complement activation. Which one of the following autoantibodies seems to play a pathogenic role in the development of membranous nephropathy?
A.
ANA
B.
Ab to megalin
C.
Ab to neutral endopeptidase
D.
Ab to phospholipase A2 receptor
E.
C and D
The answer is E
There is sufficient evidence to suggest that idiopathic membranous nephropathy (MN) is an autoimmune disease. The search for target antigens is currently underway. Studies in rats initially showed that the podocyte is the target of injury. A glycoprotein , known as megalin present on podocytes, functions as a multiligand receptor for the uptake of a variety of macromolecules such advanced glycation end products, aminoglycosides, vitamin D, etc. An Ab to megalin along with megalin has been extracted in immune complexes at the foot processes of podocytes. Formation of megalin–antimegalin Ab results in activation of the complement with insertion of C5b-9 membrane attack complex to the podocyte membrane on the urinary side of the basement membrane. The net result is excess cytokine production, oxidative stress, and growth factors, which damage the basement membrane and cause increased glomerular permeability, proteinuria, and effacement of foot processes. Although megalin appears to be a potentially important antigen, it is not present on podocytes in human subjects. Thus, megalin is an unlikely antigen in human MN.
Another antigen, neutral endopeptidase, NEP , was identified on podocytes. Ronco and associates reported MN in neonates with nephrotic syndrome and AKI from three families. They demonstrated anti-NEP-Abs in the serum of these neonates, and proposed that these antibodies were transplacentally transferred from the mother to the child that caused MN. Further analysis showed that the mother had a deficiency of NEP and probably had become immunized against the antigen at the time of or after an early miscarriage. Thus, NEP has been identified as an antigen for the development of MN.
Recently, Beck et al. identified another target antigen called M-type phospholipase A 2 receptor (PLA 2 R) , which is expressed on podocytes. Anti-PLA 2 R-Ab belongs to the IgG4 subclass of the immunoglobulins. Both PLA 2 R and its autoantibody were isolated from immune complexes of patients with MN. These autoantibodies are specific to idiopathic MN, as these antibodies were detected in 70 % of patients with idiopathic MN and were absent in patients with secondary MN. The option E is correct because both NEP and PLA 2 R are currently proven target antigens for development of idiopathic MN.
ANA is an autoantibody detected in patients with lupus. Autoantibodies to PLA 2 R are absent in lupus MN. Therefore, option A is incorrect.
Suggested Reading
Ronco P, Debiec H. Podocyte antigens and glomerular disease. Nephron Exp Nephrol 107:e41–246, 2007.
Beck LH, Bongio RGB, Lambeau G, et al. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med 361:11–21, 2009.
27.
A 56-year-old Caucasian man with type 2 diabetes, proteinuria (2 g/day), eGFR of 54 mL/min, and blood pressure of 136/84 mmHg sees you in your office for evaluation of his medications and advice for management of his kidney disease. His medications include hydrochlorothiazide (HCTZ) (12.5 mg), diltiazem CD (240 mg), losartan (100 mg) and lipitor (40 mg). Keeping evidence-based practice in mind, which one the following changes you would make to reduce proteinuria in this patient?
A.
Discontinue diltiazem CD and start Nifedipine XL 60 mg to improve BP
B.
Continue the current antihypertensives and advice the patient to consume 8 g of NaCl in the diet
C.
Continue HCTZ, diltiazem CD but switch from losartan to telmisartan (80 mg/day)
D.
Substitute chlorthalidone for HCTZ and continue other medications
E.
Reevaluate medications in 1 year
The answer is C
Several studies have shown beneficial effects of ACE-Is or angiotensin-receptor blockers (ARBs) in reducing proteinuria in both type 1 and type 2 diabetic patients . Also, thiazide diuretics (HCTZ or chlorthalidone) and nondihydropyridine calcium blockers (long-acting diltiazem or verapamil) were found to be beneficial in type-2 diabetic patients with proteinuria.
A recent multicenter trial evaluated the superiority of telmisartan, an ARB, over another ARB, losartan in type 2 diabetic patients with a mean albumin:creatinine ratio of approximately 1400 mg/g, eGFR of 50 mL/min and blood pressure >130/80 mmHg. The primary endpoint was the difference in the urinary albumin to creatinine ratio between the groups at 52 weeks. Although the reduction in blood pressure was similar in both groups, the mean reduction in albuminuria was greater in the telmisartan than in the losartan group. Thus, this study found superiority over losartan in reducing albuminuria in type 2 diabetic patients with hypertension and CKD. Thus, evidence-based medicine suggests switching losartan to telmisartan.
Suggested Reading
Bakris G, Burgess E, Weir M, et al. on behalf of the AMADEO Study Investigators. Telmisartan is more effective than losartan in reducing proteinuria in patients with diabetic nephropathy. Kidney Int 74:364–369, 2008.
28.
You are asked by a primary care physician to see a type 1 diabetic patient with normoalbuminuria (10 mg/day). He has been on enalapril 20 mg/day. His BP is 122/72 mmHg. His glycated HgbA1c is 7.5 %. The primary care physician seeks your advice for change of enalapril to losartan 50 or 100 mg/day in view of clinical trials that showed a renal benefit in type 2 diabetic patients. In view of the recent study, which one of the following suggestions you recommend to the primary care physician?
A.
Switch enalapril to losartan
B.
Continue enalapril
C.
Add losartan to enalapril
D.
Change enalapril to another ACE-I
E.
Discontinue enalapril as BP is good
The answer is B
Several studies have shown that inhibition of renin-angiotensin I-aldosterone system in type 1 and type 2 diabetic subjects by ACE-Is or ARBs is renoprotective. Most of these studies have been done in patients with micro- or macroalbuminuria with or without hypertension. However, studies in patients with normoalbuminuria and normotension have been sparce. Recently, Mauer et al. reported such a study in type 1 diabetic subjects. They conducted a multicenter, controlled trial of 285 normotensive and normoalbuminuric type 1 diabetic subjects. They were divided into three groups. One group received enalapril (20 mg daily), an ACE-I, the second group losartan (100 mg daily), an ARB, and the third group did not receive any medication and served as placebo. Both groups were followed for 5 years. The primary end point was a change in the fractional mesangial volume per glomerulus over a 5-year-period in renal biopsy specimens done before and after the study. Also, progression of retinopathy was evaluated. The results showed that the mesangial fractional volume per glomerulus did not differ significantly between the placebo and treated groups. However, albuminuria increased in the losartan group but not in enalapril group as compared with the placebo group. However, the progression of retinopathy was reduced in the treated groups as compared with the placebo group. Thus, early blockade of the renin-angiotensin-aldosterone system in type 1 diabetic subjects did not slow nephropathy progression but slowed the progression of retinopathy.
From the above discussion, it appears that continuation of enalapril is appropriate in the patient (option B is correct), and other options are incorrect. The combination of ACE-I and ARB is not suggested at this time in view of a large recent trial, using a combination of ramipril and telmisartan (the ONTARGET study).
Suggested Reading
Mann JFE, Schmieder RE, McQueen M, et al. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicenter, randomized, double-blind, controlled trial. Lancet 372:547–553, 2008.
Mauer M, Zinman B, Gardiner R, et al. Renal and retinal effects of enalapril and losartan in type-1 diabetes. N Engl J Med 361:40–51, 2009.
29.
A 48-year-old man with recently diagnosed type 2 diabetes sees his primary care physician for new onset lower extremity edema. His medications include glipizide 5 mg, lisinopril 20 mg, and HCTZ 12.5 mg. He has no retinopathy. Lungs are clear. There is no S3. Lower extremities show 3+ edema. HgbA1c is 7 %. He is referred to you for a renal biopsy. Which one of the following is NOT an indication for renal biopsy in a diabetic patient ?
A.
Proteinuria or nephrotic syndrome of sudden onset, appearing less than 5–10 years of type I diabetes
B.
Proteinuria and/or impaired renal function in the presence of retinopathy mostly in type 1 diabetes
C.
Proteinuria associated with a nephritic syndrome characterized by micro- or macrohematuria, renal insufficiency with RBC casts in types 1 and 2 DM
D.
Unexplained renal failure with or without proteinuria
E.
Presence of a systemic disease with abnormal serologic findings and clinical renal disease
The answer is B
Occasionally, patients with established diabetes present with dipstick positive or heavy proteinuria that is not expected for the duration or control of hyperglycemia. Also, unexpected deterioration in renal function is seen. Such atypical presentation is more common in type 2 diabetic than type 1 diabetic patients. These patients require a thorough clinical workup of proteinuria. Such patients usually undergo a renal biopsy to establish diabetic nephropathy and its various stages, but also to document existence of a nondiabetic renal disease alone or superimposed on diabetic nephropathy. In addition to a definitive diagnosis, the histopathologic findings provide pertinent prognostic information as well as a direction for appropriate therapy and management. The indications for renal biopsy are shown in the following table (Table 2.4 ).
Table 2.4
Indications for kidney biopsy in diabetic patients with renal disease
1. Proteinuria or nephrotic syndrome of sudden onset, appearing less than 5–10 years of type I diabetes |
2. Proteinuria and/or impaired renal function in the absence of retinopathy in type 1 diabetesa |
3. Proteinuria associated with a nephritic syndrome characterized by micro- or macrohematuria, renal insufficiency with RBC casts in types 1 and 2 diabetes |
4. Unexplained renal failure with or without proteinuria |
5. Presence of a systemic disease with abnormal serologic findings and clinical renal disease |
6. Abnormal imaging studies such as ultrasonography and doppler studies, after excluding reno-vascular disease |
7. Absence of urologic disease or infection |
When renal biopsies were performed in patients with atypical presentation, as shown in the table, a variety of nondiabetic renal diseases were observed. Such renal lesions were present either alone or superimposed on diabetic nephropathy, making management more difficult. The following glomerular lesions have been documented .
Minimal change disease /Focal segmental glomerulosclerosis
Membranous glomerulopathy
C rescentic glomerulonephritis (GN)
Post-infectious GN
IgA nephropathy-primary/secondary
Lupus nephritis
HCV associated GN
Fibrillary GN
Monoclonal immunoglobulin-mediated diseases
Collapsing glomerulopathy
It is, therefore, suggested that the nephrologist should include nondiabetic renal lesions in the differential diagnosis of abnormal proteinuria. It is believed that type 1 diabetic patients with retinopathy will have nephropathy, and renal biopsy is usually not indicated. Thus, option B is correct.
Suggested Reading
Reddi AS. Pathology of the kidney in type 1 diabetes. In Diabetic Nephropathy: Theory & Practice. East Hanover, College Book Publishers, LLC, 2004; pp. 109–129.
Seshan SV, Reddi AS. Albuminuria-proteinuria in diabetes mellitus. In Lerma EV, Batuman V (eds): The Kidney in Diabetes, New York, Springer, 2014, pp. 107–117.
30.
Which one of the following agents is MOST likely to cause the progression of kidney disease?
A.
Pirfenidone
B.
Hepatocyte growth factor
C.
Transforming growth factor-β1
D.
ACE-inhibitors
E.
Relaxin
The answer is C
Several antifibrotic agents have been tried to prevent fibrosis in the kidney and thus progression of the kidney disease in both diabetic and nondiabetic individuals. These agents include pirfenidone, hepatocyte growth factor, ACE-inhibitors and relaxin. Transforming growth factor-β1 (TGF-β1) is a cytokine that has been shown to cause fibrosis in the kidney, and anti-TGF-β1 treatment ameliorates kidney disease in animal models of diabetes .
Suggested Reading
Coppo R, Amore A. New perspectives in treatment of glomerulonephritis. Pediatr Nephrol 19:256–265, 2004
Negri AL. Prevention of progressive fibrosis in chronic renal diseases: antifibrotic agents. J Nephrol 17:496–503, 2004.
Samuel CS, Hewitson TD. Relaxin in cardiovascular and renal disease. Kidney Int 69:1498–1502, 2006.
Shihab FS. Do we have a pill for renal fibrosis? Clin J Amer Soc Nephrol 2:876–878, 2007.
Mathew A, Cunard R, Sharma K. Antifibrotic treatment and other new strategies for improving renal outcomes. Contrib Nephrol 170: 217–27, 2011.
Kania DS, Smith CT, Nash CL, et al. Potential new treatments for diabetic kidney disease. Med Clin N Am 97:115–34, 2013.
31.
A 48-year-old woman with long-standing type 2 diabetes is referred to a nephrologist for evaluation of CKD (eGFR 58 mL/min), hematuria and chronic flank pain. On history, she had frequent UTIs with E. coli infection and an episode of decreased urine output secondary to urinary tract obstruction. The patient is not ill and has no dysuria. A renal biopsy shows macrophages with abundant, foamy, and multinucleated giant cells with owl eye appearance and positive PAS staining. The history and renal biopsy findings are MOST consistent with which of the following diagnoses?
A.
Acute pyelonephritis
B.
Xanthogranulomatous pyelonephritis
C.
Malakoplakia
D.
Active renal tuberculosis (TB)
E.
Diabetic nephropathy
The answer is C
Acute pyelonephritis presents with nausea, vomiting, fever, flank or colicky pain, dysuria and frequent urination. Generally, patients are volume depleted with low BP and always ill-looking. The patient presented in the case is not ill and did not present with any one of the above clinical manifestations. Therefore, option A is incorrect.
The diagnosis of xanthogranulomatous pyelonephritis is a good possibility; however, the renal biopsy findings are not consistent with this disease. Also, the renal pathology is not consistent with diabetic nephropathy.
Renal TB is the most common extrapulmonary TB, which remains clinically silent for many years with irreversible renal damage. Acutely, urogenital TB may present with dysuria, hematuria, flank pain and pyuria. Unexplained sterile pyuria and hematuria in a clinically silent patient should prompt the clinician to consider renal TB in the differential diagnosis. Pathologic changes in the kidney include granulomatous inflammation with papillary necrosis. The patient presented in the case does not seem to carry the diagnosis of renal TB.
Based upon the history and renal pathology, the patient carries the diagnosis of malakoplakia, which is a rare granulomatous disease with many clinical similarities to xanthogranulomatous pyelonephritis. Malakoplakia is common in middle-aged women with chronic UTI. It is caused by enteric bacteria and affects many organs but frequently the urinary tract. The gross lesion appears as a soft yellow–brown plaque of variable size. Histologically, the plaques show large macrophages with foamy cytoplasm. The cytoplasm contains PAS-positive granules and crystals called Michaelis-Gutmann bodies . These bodies contain calcium and iron. Malakoplakia seems to be due to a defect in macrophage function that blocks the lysosomal enzymatic degradation of engulfed bacteria and accumulate in the cytoplasm as bacterial debris. E. coli is the most common organism cultured in the urine. Malakoplakia is commonly seen in immunosuppressive patients. Treatment with antimicrobial agents for UTI and cholinergic agonist, bethanechol, has been proven beneficial in some patients .
Suggested Reading
Diwakar R, Else J, Wong W, et al. Enlarged kidneys and acute renal failure-why is renal biopsy necessary for diagnosis and treatment. Nephrol Dial Transplant 23:401–403, 2008.
Kobayashi A, Utsunimiya Y, Kono M, et al. Malakoplakia of the kidney. Am J Kidney Dis 51:326–330, 2008.
32.
Nephrotoxicity is one of the serious side effects of antiretroviral therapy. Which one of the following statements regarding the mechanisms of renal injury is FALSE?
A.
Increase in intracellular concentration of antiretroviral drugs through human organic anion transporter 1-controlled mechanisms in the tubule
B.
Apoptosis (programmed cell death) via the mitogen-activated protein kinase (MAPK) pathway
C.
Mitochondrial damage disrupting fatty acid oxidation and energy production
D.
Crystal deposition and vascular injury
E.
Decreased renal blood flow and glomerular hypertension
The answer is E
Antiviral drug-induced nephrotoxicity is a major problem in HIV/AIDS patients. The mechanisms by which these drugs cause nephrotoxicity include (1) transport defects; (2) apoptosis; (3) mitochondrial injury; and (4) crystal deposition and vascular injury.
Basically, human organic anion transporter 1 (hOAT1) , human organic cation transporter 1 (hOCT1) , and multidrug resistance associated protein type 2 (MRP2) controlled mechanisms mediate renal tubular uptake and excretion of antiviral drugs. When these mechanisms are defective, the intracellular concentration of some antiviral drugs increases, causing tubular dysfunction. Acyclic nucleotide phosphonates (cidofovir and adefovir) toxicity is due to transport defects.
Apoptosis (programmed cell death) can occur in concert with immune-mediated injury via activation of the MAPK pathway, resulting in barrier dysfunction in renal epithelial cells. Interferon causes renal dysfunction through this pathway.
Drugs such as nucleoside reverse transcriptase inhibitors (ziduvudine, didanosine, stavudine) enter mitochondria by cellular diffusion and/or transport mechanisms and disturb respiratory-chain enzymes or mitochondrial DNA. This results in oxidative stress with ensuing anaerobic metabolism, lactic acidosis, decreased energy production, and triglyceride accumulation (microvascular fat within cells). Drugs such as acyclovir or indinavir can form crystals in renal tubules and cause AKI by intratubular obstruction. Interferon and valacyclovir have been shown to cause thrombotic microangiopathies . Changes in renal hemodynamics such as decreased renal blood flow and glomerular hypertension have not been studied in patients on antiviral drugs. Therefore, option E is incorrect.
Suggested Reading
Daugas E, Rougier J-P, Hill G. HAART-related nephropathies in HIV-infected patients. Kidney Int 67:393–403, 2005.
Izzedine H, Launay-Vacher V, Deray D. Antiviral drug-induced nephrotoxicity. Am J Kidney Dis 45:804–817, 2005.
Berns JS, Kasbekar N. Highly active antiretroviral therapy and the kidney: An update on antiretroviral medications for nephrologists. Clin J Am Soc Nephrol 1:117–129, 2006.
Jao J, Wyatt CM. Antiretroviral medications: Adverse effects. Adv Chronic Kidney Dis 17:72–82, 2010.
33.
A 30-year-old man with HIV-associated nephropathy (HIVAN) is on maintenance hemodialysis. Which one of the following drugs needs dose reduction in the patient?
A.
Norvir (Ritonavir, a protease inhibitor)
B.
Viread (Tenofovir, a nucleotide reverse transcriptase inhibitor )
C.
Sustiva (Efavirenz, a non-nucleotide reverse transcriptase inhibitor)
D.
Fugeon (Enfuvirtide, an entry fusion inhibitor)
E.
None of the above
The answer is B
Except for viread (tenofovir), the other drugs do not require any dose adjustments in patients with CKD or on dialysis. Nucleoside/nucleotide, reverse transcriptase inhibitors primarily undergo renal elimination and the other categories of drugs are metabolized in the liver. Detailed pharmacokinetic studies, however, for fugeon have not been done in patients with low GFR, but dose adjustment is not necessary.
Suggested Reading
Daugas E, Rougier J-P, Hill G. HAART-related nephropathies in HIV-infected patients. Kidney Int 67:393–403, 2005.
Izzedine H, Launay-Vacher V, Deray D. Antiviral drug-induced nephrotoxicity. Am J Kidney Dis 45:804–817, 2005.
Berns JS, Kasbekar N. Highly active antiretroviral therapy and the kidney: An update on antiretroviral medications for nephrologists. Clin J Am Soc Nephrol 1:117–129, 2006.
34.
A 60-year-old man is admitted for nephrotic syndrome and a serum creatinine concentration of 2.6 mg/dL. A renal biopsy reveals minimal change disease. Which one of the following statements is LEAST likely in this patient when compared to a similar aged male with minimal change disease and normal renal function (creatinine 1.10 mg/dL)?
A.
I ncreased systolic blood pressure (SBP)
B.
Renal failure due to renal ischemia
C.
Proteinuria much higher despite similar glomerular disease
D.
Evidence of AKI and atherosclerosis in addition to minimal change disease
E.
Irreversible acute kidney disease (AKI)
The answer is E
The association of minimal change disease with AKI has been described much more commonly in adults than in children. Studies have shown that patients with this association were older, had a higher SBP and proteinuria, and their renal biopsies showed evidence of atherosclerosis, renal ischemic changes as well as acute tubular injury. Follow-up studies of a small number of patients showed that the AKI reversed spontaneously or in some patients after dialytic therapy. Thus, AKI is reversible (E).
Suggested Reading
Nolasco F, Cameron JS, Heywood EF, et al. Adult-onset minimal change nephrotic syndrome: A long-term follow-up. Kidney Int 29:1215–1223, 1986.
Smith JD, Heyslett JP. Reversible renal failure in the nephrotic syndrome. Am J Kidney Dis 19:201–213, 1992.
35.
An 18-year-old Caucasian male student is found to have nephrotic syndrome due to minimal change disease. He is started on steroids. Regarding the pattern of response to corticosteroids in this patient, which one of the following statements is CORRECT?
A.
No response (steroid-resistant)
B.
Primary responders without any relapse
C.
Primary responders with only one relapse in the first 6 months after initial response
D.
Initial steroid response but frequent relapses within 6 months
E.
All of the above
The answer is E
Corticosteroids are the main stay of therapy for minimal change disease . In children, the usual dose is 60 mg/m 2 /day and the response is seen in 4–6 weeks in over 90 % of patients. Therapy is continued for 6 weeks after complete remission with an alternate day or reduced dosage of prednisone. There is no relapse in complete responders. However, some patients may have one or more relapses in 6 months following total or sudden withdrawal or reduction in prednisone dosage. Some of these patients with relapses may respond to regular dose of prednisone or some may not even respond to prednisone.
Some children do not respond to a 4–6 week therapy of prednisone. These children are considered steroid- resistant , and renal biopsy is indicated to consider another glomerular disease. Thus, different patterns of response to prednisone can be seen in patients with minimal change disease.
In adults, the dose of prednisone is 1 mg/kg body weight, not to exceed 80 mg/day. Unlike in children, the treatment in adults may take up to 15 weeks to observe a response.
Suggested Reading
Nachman PH, Jennette JC, Falk RJ. Primary glomerular diseases. In Taal MW, Chertow GM, Marsden PA, et al. (eds): Brenner & Rector’s The Kidney, 9th ed, Philadelphia, Elsevier Saunders, 2012, pp. 1100–1191.
Schnaper HW, Kopp JB. Nephrotic syndrome and the podocytopathies: Minimal change nephropathy, focal segmental glomerulosclerosis, and collapsing glomerulopathy. In Coffman TM, Falk RJ, Molitoris BA, et al. (eds) Schrier’s Diseases of the kidney 9th ed, Philadelphia, Wolters Kluwer/Lippincott Williams & Wilkins, 2013, pp. 1414–1521.
36.
A 36-year-old woman with rheumatoid arthritis for several years is admitted for hematuria, proteinuria, and worsening renal function. Her medications include prednisone, methotrexate and an ACE-inhibitor. Her arthritis was controlled for 8 years, but joint pain became worse 6 months ago. At that time, she was started on etanercept (TNFα-antagonist) and her symptoms improved. Her serum creatinine was 0.8 mg/dL. She had no either hematuria or proteinuria at start of etanercept. On admission, her serum creatinine is 6.2 mg/dL. ANA and anti-double-stranded DNA are positive. Urinalysis reveals 2+ blood, 4+ proteinuria, RBCs and RBC casts. Spot urine protein to creatinine ratio is 8. Which one of the following is the MOST likely diagnosis in this patient?
A.
ACE-inhibitor-induced acute kidney injury (AKI)
B.
Amyloidosis
C.
Glomerulonephritis induced by etanercept
D.
Glomerulonephritis due to long-term rheumatoid arthritis
E.
Methotrexate-induced chronic tubulointerstitial disease (TID)
The answer is C
The presence of RBCs, RBC casts and AKI are consistent with glomerulonephritis with endocapillary (endothelial and mesangial cells) proliferation. ACE-inhibitors may cause AKI in patients with bilateral renal artery stenosis or in those with decreased effective arterial blood volume. In addition, they may rarely cause membranous nephropathy. However, the clinical picture of the patient is not consistent with ACE-inhibitor use. Amyloidosis with nephrotic syndrome develops in patients with rheumatoid arthritis. However, the findings on urinalysis are not consistent with amyloidosis. Similarly, glomerulonephritis (GN) can occur in rheumatoid arthritis, but the renal course is atypical. Also, the occurrence of nephrotic syndrome in chronic TID due to methotrexate is unusual. Thus, options A, B, D and E are incorrect.
It has been shown that TNFα-antagonists (etanercept, adalimumab and infliximab) lead to formation of antibodies, including ANA, anti-double-stranded DNA and anticardiolipin antibodies in some patients with rheumatoid arthritis, resulting in reversible lupus-like syndrome. Stokes and colleagues reported their data on 5 patients with long-standing rheumatoid arthritis who developed new onset glomerular disease, proteinuria >1 g/day, and AKI. Renal biopsies showed lupus nephritis (classes III and IV) in 2 patients, pauci-immune necrotizing crescentic GN in 1 patient, pauci-immune necrotizing crescentic GN and amyloidosis in 1 patient, and membranous nephropathy and renal vasculitis in another patient. Median duration of therapy was 6 months (3–30 months). Similarly, Chin et al. described a psoriatic patient who developed nephrotic syndrome due to membranous nephropathy following infliximab administration. In both studies, discontinuation of TNFα-antagonists improved proteinuria and renal function . Thus, the use of TNFα-antagonists may induce renal disease in some patients. Since our patient developed renal disease and lupus serology in 6 months following therapy with etanercept, option C is the correct answer.
Suggested Reading
Chin G, Luxton G, Harvey JM: Infliximab and nephrotic syndrome. Nephrol Dial Tansplant 20:2824–2826, 2005.
Stokes MB, Foster K, Markowitz GS, et al., Development of glomerulonephritis during anti-TNF-α therapy for rheumatoid arthritis. Nephrol Dial Tansplant 20:1400–1406, 2005.
De Bandt: Lessons for lupus from tumor necrosis factor blockade. Lupus 15:762–767, 2006.
37.
Which one of the following conditions is NOT associated with FSGS?
A.
Morbid obesity
B.
IgG 4
C.
Sickle cell disease
D.
HIV infection
E.
Low nephron number
The answer is B
Except for IgG 4, which causes interstitial nephritis , all other conditions are associated with FSGS and nephrotic syndrome . FSGS in obesity and decreased number of nephrons may result from intraglomerular hypertension, whereas that in sickle cell anemia it may be due to hypoxemia. How HIV infection causes FSGS is not clearly understood, however, the involvement of NEF gene has been implicated.
Suggested Reading
Nachman PH, Jennette JC, Falk RJ. Primary glomerular diseases. In Taal MW, Chertow GM, Marsden PA, et al. (eds): Brenner & Rector’s The Kidney, 9th ed, Philadelphia, Elsevier Saunders, 2012, pp. 1100–1191.
Schnaper HW, Kopp JB. Nephrotic syndrome and the podocytopathies: Minimal change nephropathy, focal segmental glomerulosclerosis, and collapsing glomerulopathy. In Coffman TM, Falk RJ, Molitoris BA, et al. (eds) Schrier’s Diseases of the kidney 9th ed, Philadelphia, Wolters Kluwer/Lippincott Williams & Wilkins, 2013, pp. 1414–1521.
38.
Get Clinical Tree app for offline access
Which one of the following statements is FALSE regarding the long-term renal survival in patients with FSGS?
A.
Non-nephrotic range proteinuria correlates with better renal survival than massive proteinuria (>10 g/day)
B.




Renal survival is worse in patients who had remission of nephrotic syndrome than those without remission of their nephrotic syndrome
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:




Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
