Chapter 1.9
Gastrointestinal tract and appetite control
Jaimini Cegla and Stephen R. Bloom
Imperial College London, London, UK
Despite fluctuations in food intake and physical activity, healthy adults maintain a relatively constant weight over decades. However, as stated by the laws of thermodynamics, if less energy is expended than consumed then the excess energy will be stored. It is calculated that an average North American man will increase his weight by 9.1 kg between 25 and 35 years of age, as a consequence of a mere 0.3% imbalance between energy consumed and energy expended over this period [1]. It is therefore of no surprise that, as a result of readily available high-energy food and our sedentary lifestyles, obesity has become a growing global epidemic. The converse is true in disorders that culminate in reduced energy intake such as anorexia nervosa. An understanding of the mechanisms that control body weight, by co-ordinating food intake and energy expenditure, is key in unravelling the pathogenesis of disordered energy homeostasis in gastrointestinal disease.
1.9.1 Role of the gut neuroendocrine system in appetite regulation
Several neural, hormonal and psychological factors control the complex process known as appetite. The hypothalamus and brainstem receive these peripheral neural and hormonal signals and co-ordinate a response in order to achieve energy homeostasis. Two discrete populations of neurones present in the arcuate nucleus (ARC) of the hypothalamus with opposing effects on food intake are crucial in this process: medially located orexigenic neurones (i.e. those stimulating appetite) express neuropeptide Y (NPY) and agouti-related peptide (AgRP), and anorexigenic neurones (i.e. those inhibiting appetite) in the lateral ARC express pro-opiomelanocortin (POMC) and cocaine and amphetamine-regulated transcript (CART) [2]. Additionally, ARC neurones also project onto the paraventricular nucleus (PVN) of the hypothalamus where important efferent pathways regulating energy expenditure arise.
The important inputs to this intricate neural network are twofold. Firstly, the short-term signals that govern meal ingestion are primarily regulated by the ‘gut–brain axis’ and secondly, information regarding long-term energy stores is signalled via leptin, an adipose-derived hormone [3]. This ‘gut–brain axis’ exists to contribute to the short-term feelings of satiety and hunger, by transmitting information from the gastrointestinal tract to the hypothalamus and brainstem, via gut hormones and the vagus nerve (see Figure 1.9.1). The majority of these gut hormones are anorexigenic and include peptide tyrosine- tyrosine (PYY), pancreatic polypeptide (PP), glucagon-like peptide-1(GLP-1), oxyntomodulin (OXM) and cholecystokinin (CCK). The only truly orexigenic hormone to be discovered thus far is ghrelin. These hormones act in concert as meal initiators and terminators [4].
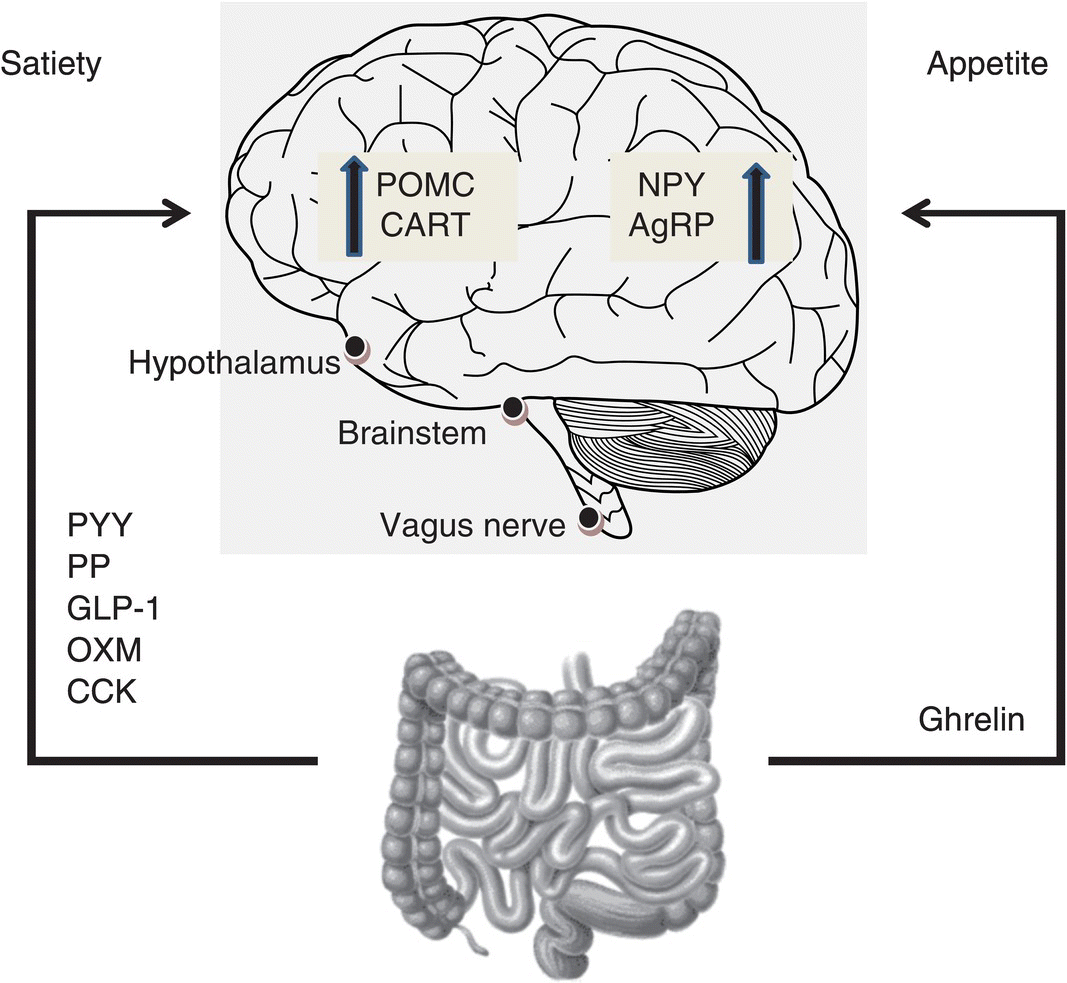
Figure 1.9.1 Mechanism of gut hormone action. Hormones released from the gastrointestinal tract into the circulation act on the brain to modulate appetite. Three sites are known to be of key importance: the hypothalamus, brainstem and vagus nerve. AgRP, agouti-related peptide; CART, cocaine- and amphetamine-regulated transcript; CCK, cholecystokinin; GLP-1, glucagon-like peptide 1; NPY, neuropeptide Y; POMC, pro-opiomelanocortin; OXM, oxyntomodulin; PP, pancreatic polypeptide; PYY, peptide tyrosine-tyrosine.
These endogenous gut hormones act on the central nervous system either via the circulation through areas deficient in the blood–brain barrier, such as the median eminence of the hypothalamus and the area postrema [5] or via receptors of vagal afferents, together with stretch receptors and nutrient chemoreceptors [6]. These signals converge in the nucleus of the tractus solitarius (NTS) of the brainstem and are integrated with information from higher brain centres relaying reward drive and mood to regulate appetite and control energy expenditure. In this chapter, a review of the key gut hormones implicated in appetite regulation is undertaken.
1.9.2 Peptide tyrosine-tyrosine
Peptide tyrosine-tyrosine is a member of the PP-fold family of peptides, which also includes the anorexigenic PP and the orexgenic neurotransmitter NPY, all sharing a common tertiary structure. PYY exists endogenously in two forms: peptide tyrosine-tyrosine1–36 and PYY3–36 [7]. Enzymatic cleavage of secreted PYY1–36 at the amino terminal by the enzyme dipeptidyl peptidase IV (DPP-IV) gives rise to PYY3–36 [8], the predominant form of circulating PYY.
Peptide tyrosine-tyrosine is released postprandially by the L-cells of the distal gut in proportion to the energy content consumed. Plasma PYY3–36 concentrations rise within 15 min of food ingestion, well before nutrients reach the colon or rectum where it is released, implicating a neural or hormonal mechanism for its release [9]. PYY3–36 concentrations peak 1–2 h postprandially and remain elevated for up to 6 h [10]. Protein-rich meals cause the greatest increase in PYY concentrations compared to other macronutrients [11]. The effects of PYY are thought to be mediated centrally via the G-protein-coupled Y receptors, in particular the Y2 receptor [12], which is densely expressed in the ARC. Peptide tyrosine-tyrosine may also exert its actions via the vagus nerve which also expresses the Y2 receptor [13]. Interestingly, obese individuals have lower fasting PYY3–36 concentrations than their lean counterparts, suggesting that obesity is a ‘PYY-deficient’ state [10].
Peptide tyrosine-tyrosine changes occur in several gastrointestinal disorders. PYY concentration in tissue extracts from the ileum and colon of patients with Crohn’s colitis and ulcerative colitis has been found to be lower than in controls [14]. Furthermore, rectal and fasting plasma PYY concentrations have been reported to be reduced in patients with ulcerative colitis. This decrease in PYY could contribute to the development of the symptoms seen in inflammatory bowel disease; for example, diarrhoea may be brought about by hindering the ileal brake and loss of inhibition of intestinal transit. Conversely, total PYY concentrations have recently been found to be increased, both pre- and postprandially, in patients with Crohn’s disease affecting the small intestine. It appears that enhanced enteroendocrine cell responses may play a role in feeding disturbance and weight loss [15]. In patients with coeliac disease, basal and postprandial plasma concentrations of PYY are elevated. These elevated concentrations of PYY normalise within 8 months on a gluten-free diet [16]. PYY concentrations in cerebrospinal fluid (CSF) have been found to be elevated in normal-weight bulimic patients abstinent for a month from pathological eating behaviour. However, CSF PYY concentrations were not affected in women who had recovered from anorexia [17]. Plasma concentrations of PYY have not yet been investigated in this group of patients.
1.9.3 Pancreatic polypeptide
Pancreatic polypeptide is an amidated 36-amino acid peptide, structurally similar to PYY. It is released postprandially under vagal control from pancreatic islet PP cells, in proportion to caloric intake. A biphasic response is observed postprandially and concentrations remain elevated for up to 6 h after a meal [18]. Pancreatic polypeptide is thought to mediate its effects via the Y4 receptor, which is highly expressed in the brainstem and ARC [19].
Fasting plasma concentrations of PP are lower in obese individuals, as is the second phase of release after food consumption [20]. Children with Prader–Willi syndrome, a condition characterised by hyperphagia and obesity, have reduced fasting and postprandial concentrations of PP [21]. In contrast, increased concentrations of PP are observed in patients with anorexia nervosa [22]. Pancreatic polypeptide release is reduced in patients with slow transit constipation, but increased in those with functional diarrhoea [23]. Levels are unchanged in patients with coeliac disease [24] but patients with ulcerative colitis have significantly elevated fasting PP concentrations [25].
1.9.4 Glucagon-like peptide 1
Glucagon-like peptide 1 is a product of proglucagon, secreted by enteroendocrine L-cells in the intestine. After a meal, it is released in response to direct L-cell stimulation by nutrients within the GI lumen, and indirectly via neuronal pathways within the enteric nervous system. Glucagon-like peptide 11-37 is processed intracellularly to generate the 7–37 and 7–36 amide peptides. Glucagon-like peptide 1 is inactivated by dipeptidyl peptidase-4 (DPP-4) which processes GLP-17-37 and GLP-17-36 amide to GLP-19-36 amide.
Glucagon-like peptide 1 binds the G-protein-coupled GLP-1 receptor expressed by pancreatic islet cells as well as brain, heart and lung tissue [26]. The actions of GLP-1 are best characterised in the beta cell where GLP-1 exerts an incretin effect, the stimulation of glucose-dependent insulin release from pancreatic beta cells [27]. Therefore, until recently, the focus on GLP-1 has been largely as an antidiabetic agent and two long-acting analogues of GLP-1, exenatide and liraglutide, are licensed for the treatment of diabetes.
In contrast to insulin, however, GLP-1 causes a decrease in body weight [28]. Acute intravenous injection of GLP-1 reduces appetite and energy intake [29]. This effect has been observed in lean, obese and diabetic volunteers. The actions of GLP-1 on appetite are likely to be related to a direct effect on the central nervous system (CNS) via activation of POMC-expressing neurones in the ARC, as well as through delayed gastric emptying [30].
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


