Gastrointestinal Neuroendocrine Lesions
General Organization of the Gastrointestinal System
The gastrointestinal neuroendocrine system (GNES) is the largest and most complex endocrine organ in the body (1). The gut contains numerous neuroendocrine (NE) cells that produce many peptide hormones. Since NE cells exhibit en-docrine, paracrine, and neurotransmitter functions (Fig. 17.1), they are best termed NE cells rather than endocrine cells. They constitute a complex system that regulates many gastrointestinal (GI) functions. Some NE secretions act as true peptide hormones. Gastrin, secretin, and cholecystokinin (CCK) are secreted into the blood to reach their target organs (stomach, pancreas, and gallbladder); soon thereafter they are metabolized and eliminated. Other peptides, such as somatostatin, are released into the local subepithelial connective tissue or directly onto other cell types via long basal cytoplasmic processes in a paracrine fashion. Neurons interact with gastrointestinal endocrine cells, endocrine cells interact with other endocrine cells, and endocrine cells may influence neurons (1,2). In addition, many GI hormones interact with the hypothalamic-pituitary axis to orchestrate the secretory activity and motility necessary for effective digestion (1), including acid, bicarbonate, and enzyme secretion and local blood flow. The GNES also interacts with the immune system (1). Immune responses alter neural and endocrine function, and in turn, neural and endocrine activity modifies immunologic functions. Finally, some GI hormones enhance metabolic levels and promote GI growth.
NE cells are widely distributed throughout the epithelia of the stomach, intestines, distal esophagus, and anus. Their overall density, contents, and structure differ in various parts of the gut. Most NE cells lie within the epithelium, but some are also present in the lamina propria of the stomach and the appendix. Endocrine cells are sensitive to chemical and mechanical stimuli, to which they respond by releasing extracellular mediators. At least 14 types of NE cells populate the GI mucosa (Table 17.1) (2). Some peptides are produced solely in the upper GI tract and are stimulated only for a short time following meals; others are scattered throughout the gut and are exposed to prolonged stimulation. Gastrin is primarily a gastric antral hormone; CCK, secretin, gastric inhibitory polypeptide, and motilin are mainly upper small intestinal hormones; and enteroglucagon and neurotensin are lower intestinal peptides. In contrast, vasoactive intestinal polypeptide (VIP), substance P, enkephalins, bombesin, somatostatin, and other substances are produced more diffusely throughout the gut.
NE cells may be open or closed, depending on whether or not they reach the gastrointestinal lumen (Fig. 17.2). Closed endocrine cells fail to reach the lumen, suggesting that they do not react to luminal stimuli. Instead, they probably react to other stimulants, such as distension, temperature, and neural or hormonal factors. “Open” cells extend from the basal lamina to the lumen. Most open endocrine cells reach the lumen in a narrow specialized area containing tufts of microvilli and a centriole that acts as a sensor of the luminal contents.
Identification of Neuroendocrine Cells
NE cells can sometimes be recognized in routinely stained sections by the presence of pyramidally shaped eosinophilic or clear cells lying along the basement membrane. Eosinophilic granules, which generally are smaller than those seen in Paneth cells, may be visible and lie diffusely distributed throughout the cell or in a subnuclear location (Fig. 17.3). Since not all NE cells are immediately recognizable in hematoxylin and eosin (H&E)-stained sections, numerous techniques have been devised to detect their presence. The earliest techniques involved reactions with various heavy metals, especially silver (Table 17.2), and ultrastructural evaluation. In this country, these techniques have largely been abandoned due to their expense, the difficulty in performing them well, and the more recent development of a number of immunostains that reliably detect endocrine cells. Immunologic markers of neuroendocrine differentiation include antibodies to neuron-specific enolase (NSE), protein gene product (PGP) 9.5, synaptophysin, and chromogranin (CgA). Of these, NSE is the least specific. CgA is a secretory granule that is located alongside specific hormones in large dense-core vesicles of neuronal and NE cells (3). It is secreted in tumors; the amount present in the blood correlates with the tumor burden. NE cells containing amines (histamine and serotonin) regularly exhibit CgA immunoreactivity.
Other peptide-containing endocrine cells are heterogeneous in their CgA immunoreactivity. We prefer synaptophysin immunostains since they reliably detect a wide variety of gastrointestinal NE cells. These antibodies and the silver-based techniques allow the identification of endocrine cells but do not provide information with respect to their hormonal products. Specific cell types can be identified immunohistochemically using antibodies directed against the specific hormone that the cells are known to produce.
Other peptide-containing endocrine cells are heterogeneous in their CgA immunoreactivity. We prefer synaptophysin immunostains since they reliably detect a wide variety of gastrointestinal NE cells. These antibodies and the silver-based techniques allow the identification of endocrine cells but do not provide information with respect to their hormonal products. Specific cell types can be identified immunohistochemically using antibodies directed against the specific hormone that the cells are known to produce.
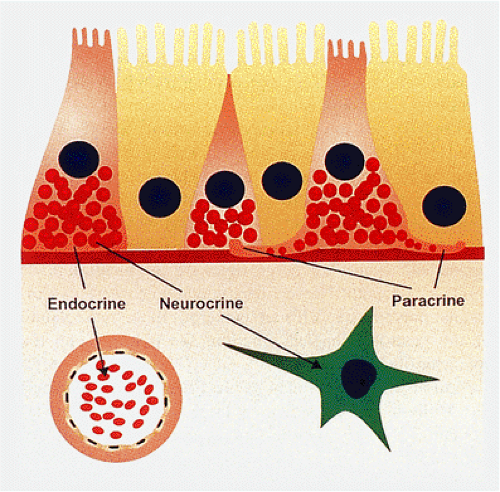 FIG. 17.1. Endocrine cells have endocrine functions whereby the contents of their secretory granules are released into the circulation to have an effect at a location distant from the site of production. They also have neurocrine effects influencing neuronal functions. Long basal extensions of endocrine cells that touch other endocrine cells or nearby epithelial cells mediate paracrine effects. |
In this chapter, we will first discuss normal gastrointestinal NE cell populations. This will be followed by a discussion of gastrointestinal NE cell proliferations. These include hyperplastic lesions as well as NE tumors.
Normal Gastrointestinal Neuroendocrine Cell Populations
Esophagus
Stomach
There are at least eight distinct gastric endocrine cell types, including enterochromaffin (EC), enterochromaffinlike (ECL),
D, D1, P, G, X, and ghrelin-producing cells (Table 17.1) (6). Three cell types (ECL, G, and D) account for >75% of the gastric NE cell mass. NE cells appear as cuboidal or short columnar cells scattered among the gastric epithelia, usually in the glands (Fig. 17.4). Less commonly they occur in the glandular neck. NE cells rarely reach the foveolae; they are absent from the surface. Their composition and distribution differ in the oxyntic and antral mucosa. Approximately 50% of antral NE cells are G cells (gastrin producing), 30% are serotonin-producing EC cells, and 15% are somatostatin-producing D cells. The remaining 5% consist of other cell types. The predominant NE cell in the fundus is the histamine-producing ECL cell. There are also a few X cells with an unknown secretory product, ghrelin-producing cells, and EC cells. Most gastric NE cells are closed. They lie along the basement membrane and are covered by nonendocrine epithelium that prevents their contact with the glandular lumen.
They contain numerous basally located hormone-containing granules. The NE cells secrete their hormones into the intercellular space, where they diffuse into capillaries. Scattered endocrine cells are occasionally found in the lamina propria near the base of glands apparently unattached to epithelium. They exist singly (Fig. 17.5) or in clusters and are seen in the antrum or antrocorpus junction. Lamina propria endocrine cells occur predominantly in the stomach of patients with gastritis, but they can also be found in the normal stomach.
D, D1, P, G, X, and ghrelin-producing cells (Table 17.1) (6). Three cell types (ECL, G, and D) account for >75% of the gastric NE cell mass. NE cells appear as cuboidal or short columnar cells scattered among the gastric epithelia, usually in the glands (Fig. 17.4). Less commonly they occur in the glandular neck. NE cells rarely reach the foveolae; they are absent from the surface. Their composition and distribution differ in the oxyntic and antral mucosa. Approximately 50% of antral NE cells are G cells (gastrin producing), 30% are serotonin-producing EC cells, and 15% are somatostatin-producing D cells. The remaining 5% consist of other cell types. The predominant NE cell in the fundus is the histamine-producing ECL cell. There are also a few X cells with an unknown secretory product, ghrelin-producing cells, and EC cells. Most gastric NE cells are closed. They lie along the basement membrane and are covered by nonendocrine epithelium that prevents their contact with the glandular lumen.
They contain numerous basally located hormone-containing granules. The NE cells secrete their hormones into the intercellular space, where they diffuse into capillaries. Scattered endocrine cells are occasionally found in the lamina propria near the base of glands apparently unattached to epithelium. They exist singly (Fig. 17.5) or in clusters and are seen in the antrum or antrocorpus junction. Lamina propria endocrine cells occur predominantly in the stomach of patients with gastritis, but they can also be found in the normal stomach.
TABLE 17.1 Gastrointestinal Endocrine Cells | |||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
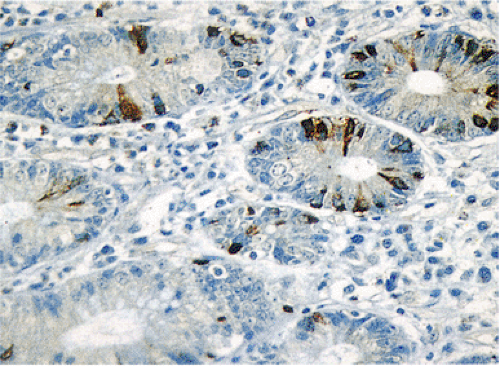 FIG. 17.2. Open and closed endocrine cells in normal colonic mucosa stained with an antibody to chromogranin. |
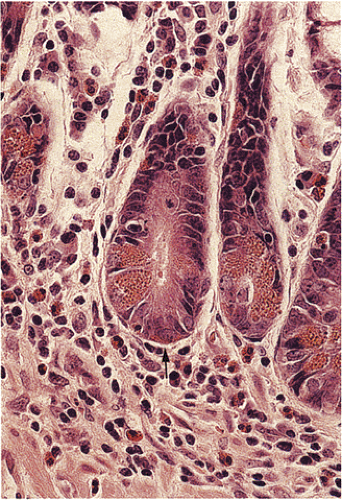 FIG. 17.3. Cross section of several crypt bases showing a mixture of cell types. The endocrine cell highlighted by the arrow lies beneath other cell types. |
TABLE 17.2 Special Stains Used to Detect Neuroendocrine Cells | ||||||
|---|---|---|---|---|---|---|
|
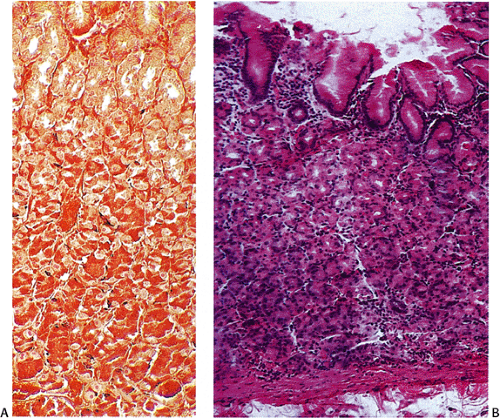 FIG. 17.4. Argyrophilic endocrine cells in the normal fundus stain with Grimelius stain (A). Corresponding hematoxylin and eosin stain (B). |
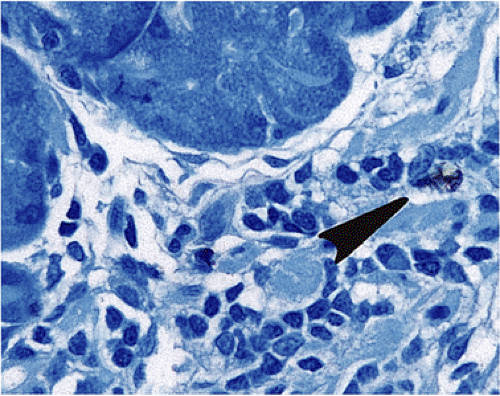 FIG. 17.5. Widely scattered endocrine cells (arrow) within the lamina propria. Chromogranin immunostain. |
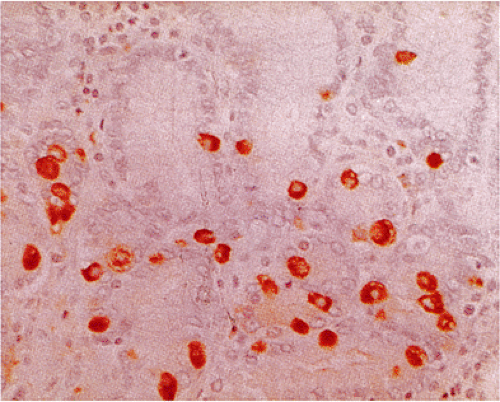 FIG. 17.6. Antral G cells stained with gastrin immunostain. |
G Cells
G cells are large, round or oval cells (7) that produce gastrin. They mainly lie in the neck region of the antral glands trailing off toward the gland base. G cells normally have an irregular and random spatial distribution that ranges between one and three to four cells per gland. They contain variably dense cytoplasmic granules measuring 150 to 200 in μm diameter. Argyrophilic and argentaffinic stains fail to detect most G cells (7), but they are easily identified by CgA or synaptophysin stains. They are also identifiable using gastrin-specific immunohistochemical stains (Fig. 17.6).
The number of antral G cells varies, depending on the acid content of the stomach, the presence of proliferative stimuli, and gastric location. G cells are most numerous on the greater curvature. Gastrin promotes acid and pepsinogen secretion (Fig 17.7), gastric motility, and gastric oxyntic mucosal proliferation. In sustained hypergastrinemia, parietal cells, ECL cells, and surface mucosal cells increase in number. The trophic effect of gastrin is most marked on ECL cells (8). Parietal cell mass and G-cell density are interrelated.
Hypochlorhydria causes G-cell proliferation and increased gastrin transcription.
Hypochlorhydria causes G-cell proliferation and increased gastrin transcription.
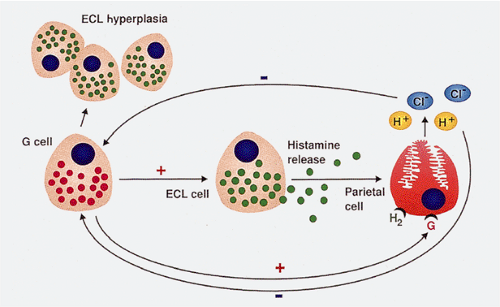 FIG. 17.7. Gastrin effects on enterochromaffinlike (ECL) cells. Gastrin release stimulates ECL hyperplasia by binding to gastrin receptors on ECL cells. This results in hyperplasia of ECL cells as well as histamine release. The histamine released from the ECL cells binds to histamine receptors on G cells leading to acid secretion. Gastrin also acts directly on parietal cells by binding to gastrin receptors present on these cells, again causing acid secretion. Acid produced by the parietal cell negatively regulates gastrin secretion through a feedback loop. |
Enterochromaffin Cells
EC cells are the most numerous NE cell populations in the gut and they are widely found in many sites including the stomach. They are sparse in the mucous neck portion but are quite numerous in the lower half of the gland. Their overall distribution is patchy; areas with numerous EC cells alternate with areas containing few or none. They are especially numerous in areas of intestinal metaplasia (9). These cells are described further below.
D Cells
D cells secrete somatostatin and are uniformly distributed throughout the antral and oxyntic mucosa (Fig. 17.8). Approximately 20% of gastric D cells have basal axonal-like cytoplasmic processes with terminal expansions that allow D cells to function in a paracrine fashion (10). Most antral D cells are open cells that act as receptors, interacting with luminal contents. A feedback mechanism exists in which intraluminal acid stimulates somatostatin secretion. In the fundus, D cells are closed. Somatostatin secretion from D cells inhibits gastric acid, gastrin and intrinsic factor secretion, and acetylcholine release.
Enterochromaffinlike Cells
ECL cells are confined to the oxyntic mucosa, where they represent the dominant NE cell type, constituting 40% to 45% of oxyntic NE cells (6). ECL cells express CCK-2 (gastrin) receptors, which mediate histamine secretion and ECL cell growth (11,12). ECL cells decrease in number after antrectomy due to reduced gastrin levels. The pyramidally shaped ECL cells are closed with a broad base that directly abuts the basement membrane of oxyntic glands. They are randomly scattered throughout the lower and middle thirds of the glands (11). Their lateral processes extend to, and terminate on, parietal cell surfaces, allowing them to act in a paracrine fashion. They also act in an endocrine fashion. ECL cells can be identified by silver stains, or more specifically, by antibodies against histamine, histidine decarboxylase, and vesicular monoamine transporter 1 and 2 (VMAT 1 and 2) (13). ECL cells are a self-renewing population.
Ghrelin-producing Cells
Ghrelin is a recently discovered peptide that is produced by about 20% of endocrine cells in the oxyntic mucosa. These cells appear to differ from the ECL, EC, D, and probably X cells also present in this area (14). Ghrelin stimulates release of growth hormone (15), stimulates food intake, and modulates sleep (16). It also has an antiproliferative effect in neoplastic tissues (17) and stimulates gastric contractility (18). The hormone localizes to polygonal or flask-shaped endocrine cells in the oxyntic mucosa. These cells appear larger than the adjacent cells (19).
Small Intestine
While NE cells constitute <1% of all intestinal epithelial cells, numerous subpopulations of NE cells are present in this site (20). NE cells lie interspersed among absorptive cells and goblet cells in the crypts (Fig 17.9) and on the villi (Fig. 17.10), and some are found within Brunner glands. They share a common origin from the crypt epithelial stem cell (21). Each subpopulation has a distinct distribution along the cranial–caudal axis and along the crypt–villus axis, thereby producing an integrated response to various stimuli and facilitating normal secretory, absorptive, and motor functions.
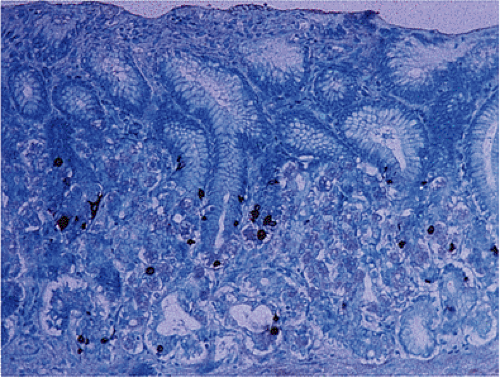 FIG. 17.8. Somatostatin-containing cells are quite numerous in the antrum. |
 FIG. 17.9. Small intestinal enterochromaffin (EC) cells stained with an antibody to serotonin. |
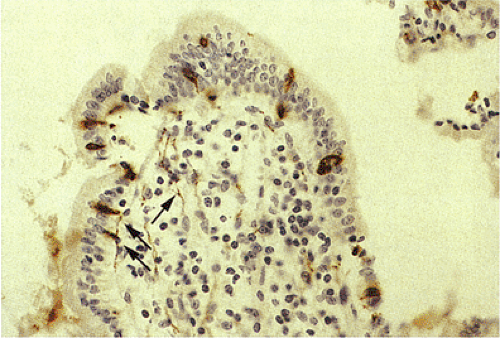 FIG. 17.10. Synaptophysin immunostain demonstrating endocrine cells along the surface of the villi, as well as numerous intramucosal nerves (arrows). |
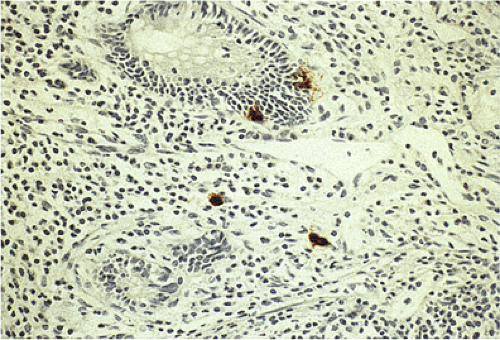 FIG. 17.11. Endocrine cells in the epithelium of the appendix. Chromogranin stain demonstrates endocrine cells in the crypt epithelium as well as in the lamina propria. |
EC cells have an almost exclusively intraepithelial location resting on the basement membrane and projecting into the lumen with their apical portions. They are evenly distributed between crypts and villi. They are recognizable by their bright red granules. Others appear clear. Their cytoplasm is occupied by a large number of secretory granules. The major secretory product is serotonin, but subsets of EC cells also produce minor amounts of tachykinins, enkephalins, and motilin (7). Serotonin is synthesized from the amino acid tryptophan by hydroxylation and decarboxylation in the EC cell cytoplasm and subsequently transported into secretory granules by an active transport mechanism. Upon specific membrane simulation, granules are translocated to the cell membrane and released by exocytosis. Serotonin may influence adjacent cells by paracrine mechanisms or reach distant cells via the circulation. Release of serotonin in response to an acid pH, hypertonic glucose, amino acids, and noxious stimuli affects nerve endings that evoke mucosal hyperemia, secretion, and peristalsis (22).
The duodenum and jejunum harbor a large number of different NE cell types; the spectrum gradually narrows as one proceeds distally (23). The duodenum contains EC cells, D cells, and cells immunoreactive for CCK and gastrin. Gastric inhibitory polypeptide–containing cells occur in the midzone of the duodenal glands and to a lesser extent in the jejunum (24). Motilin-immunoreactive cells populate the duodenum and upper jejunum. Substance P cells in the proximal small intestine occur mainly in the crypts and lower villi, whereas secretin-containing cells are found nearly exclusively on villi. Brunner glands also contain NE cells producing and storing somatostatin, gastrin, CCK, and peptide YY (25).
Appendix
The appendix contains two populations of EC cells: Those in the crypts and those in the lamina propria (Fig. 17.11). They occur singly or in small clusters, and are more common in adults than in children. Lamina propria NE cells lie scattered near the base of the crypts, apparently unattached to crypt epithelium. They are invariably part of a structure termed the enterochromaffin cell–nerve fiber complex (26) that contains a mixture of endocrine cells, neurons, Schwann cells, and unmyelinated nerve processes all surrounded by an external lamina that is continuous with that of adjacent peptidergic nerve fibers. The EC cell–nerve fiber complex facilitates integration between intestinal endocrine cells and the enteric nervous system. Crypt endocrine cells tend to lie near the base of the crypts. Both crypt and lamina propria endocrine cells contain serotonin, somatostatin, enteroglucagon vasoactive intestinal polypeptide, and substance P. More endocrine cells populate the distal appendix than the proximal appendix. ECL cells, D cells, L cells, and N cells are also present.
Large Intestine, Rectum, and Anus
The colon has the least number of NE cells of any region of the GI tract, except the esophagus. The endocrine cell population in this region is heterogeneous in nature (Table 17.1). NE cells appear as small round or pyramidal cells lying on the basement membrane and scattered among the nonendocrine epithelial cells. They are most numerous in the base of the crypts and appear clear or they contain prominent eosinophilic basal granules. The latter are released by pinocytosis from the basal and lateral surfaces of the cells. Secretory products of these cells exert a paracrine modulating effect on neighboring exocrine and endocrine cells. When these products cross the basal membrane, they enter the bloodstream reaching target organs, where they exert endocrine effects. The neural effects of these cells are ex-erted as the hormones diffuse to synapses.
Anal endocrine cells lie above the dentate line in the colorectal mucosa, in the transitional mucosa, in anal ducts and glands, in crypts, and in perianal sweat glands. Like endocrine
cells elsewhere, they lie close to the basement membrane. Endocrine cells are absent in the pectineal folds and perianal skin.
cells elsewhere, they lie close to the basement membrane. Endocrine cells are absent in the pectineal folds and perianal skin.
Molecular Features of Gastrointestinal Endocrine Tumors
Neuroendocrine tumors (NETs) exhibit a series of genetic aberrations, including point mutations, gene deletions, DNA methylation, chromosomal losses, and chromosomal gains involving both oncogenes and tumor suppressor genes. Several genetic syndromes with mutations in tumor suppressor genes, including multiple endocrine neoplasia type 1 (MEN-1) and neurofibromatosis type I (NF1), associate with the development of gastrointestinal NE tumors. MEN-1, an autosomal dominant disorder, associates with mutations in the MEN1 gene on chromosome 11q13. MEN1 encodes the protein menin, which binds Jun1, inhibiting Jun1-activated transcription (27,28). Mutations and/or loss of heterozygosity (LOH) at the MEN1 locus occur in 40% to 75% of gastrointestinal NE tumors (27,29). The frequency of this event differs depending on tumor site and whether the tumors are sporadic or arise as part of the MEN-1 syndrome. For example, foregut and some midgut NETs have frequent deletions and mutations in the MEN1 gene (30). MEN1 LOH is present in 33% of midgut carcinoid tumors (31). Type II gastric carcinoids show LOH at the MEN1 locus in 75% of tumors as compared to 16% of type I gastric carcinoids (32). LOH at the MEN1 locus appears to be an important prerequisite to switch hypergastrinemia-induced ECL proliferations from a hyperplastic to a neoplastic state (31). Further, the presence of 11q13 LOH in gastric carcinoid tumors of patients without hypergastrinemia suggests that inactivation of the MEN1 gene alone can cause ECL cell tumors (33). Reg-a gene alterations may be important in gastric ECL carcinoids (34). Mutation of the Reg-a gene could contribute to the development of ECL cell carcinoid tumors by allowing unrestrained stimulating effects of gastrin (34). Alterations in the MEN1 gene also occur in 25% to 40% of sporadic gastrinomas (35).
LOH at locations distal to 11q13 at the location of the succinate ubiquinone oxidoreductase subunit D (SDHD) tumor suppressor gene is also implicated in the development of midgut (rather than foregut) carcinoids (36). Twenty-two percent of duodenal and ileal carcinoids show alterations at the SDHD locus (36). A number of other chromosomal gains and losses develop in NETs, but losses at 18qter, 11q22-24, and 16q are the most common genetic defects in midgut carcinoids (37,38). LOH at chromosome 18q is seen in 67% of midgut tumors (37). Fifteen percent of midgut tumors show abnormalities of the X chromosome (39). Inactivation of the p16INKα/CDKN2A tumor suppressor gene on chromosome 9p21 is also common in NE tumors. p16INKα4 inactivation occurs by either gene deletion or methylation of CpG islands (40) and is observed in 50% to 52% of sporadic gastrinomas (35). Other abnormalities found in gastrinomas include aneuploidy, mismatch repair defects, amplification of the HER2/neu protooncogene (41), and other genetic changes.
Almost 80% of gastrointestinal NETs demonstrate nuclear and cytoplasmic β-catenin expression and 37.5% contain an exon 3 mutation (42). Another gene that may be important in the development of NETs is the PDCD4 (programmed cell death protein 4) gene (43). PDCD4 is a newly described tumor suppressor gene that suppresses cell proliferation and lies close to the MEN1 gene on chromosome 11q13. Sporadic endocrine tumors arising in several sites frequently show loss of its expression.
NF1 is an autosomal dominant disorder resulting from mutations in the NF1 gene located at 17q11. These mutations lead to premature truncation of neurofibromin, the tumor suppressor gene product. Some NF1 patients develop duodenal somatostatinomas (44) or gangliocytic paragangliomas. Several genes are frequently methylated in GI NETs including p14, p16, MGMT, THBS1, RARβ, ER, and COX2 (37,45). p16 methylation is more frequent in older patients and associates with metastasis (45). The Ras-association domain family 1, isoform A (RASSF1A) gene is also frequently methylated in these neoplasms and associates with lymph node metastasis (45). Allelic losses of chromosomes 11q, 16q, and 1q may be important in the pathogenesis of goblet cell carcinoids and ileal carcinoids (46).
Immunohistochemical studies describe overexpression of the antiapoptotic bcl2 protein in gastric endocrine cell hyperplasia (47) and loss of E-cadherin expression in two thirds of malignant rectal carcinoid tumors (48). p53 expression is rare in small intestinal NETs (49) but occurs in up to 16% of carcinoid tumors (50). p53 mutations are found in up to 25% of appendiceal goblet cell carcinoids as well as in some conventional appendiceal carcinoids (51). Ki-67 staining and p53 staining may serve as prognostic factors in some tumors (52,53).
Transforming growth factor-α (TGF-α) is expressed in 72% of gastrointestinal carcinoid tumors and most tumors also express its receptor, epidermal growth factor receptor (EGFR) (37). Gastrin-releasing peptide (GRP) is expressed in a proportion of NETs and gastrin-releasing peptide receptor (GRPR) is expressed in the majority of such tumors. Both GRP and GRPR are coexpressed in some gastrointestinal NETs, although GRP is not expressed by normal gastrointestinal neuroendocrine cells (54). Other proteins that are frequently expressed include c-myc, c-erbB2, and c-jun (55). These tumors also frequently express the somatostatin receptor subtype 2, a factor that may help select patients suitable for somatostatin analog treatment (56).
Neuroendocrine Cell Hyperplasia
The term NE cell hyperplasia signifies a nonneoplastic, nonautonomous proliferation (more than two times normal) of NE cells leading to increased numbers of NE cells per unit area and increases in their total cell mass (57). It develops in
various chronic inflammatory conditions, including chronic atrophic gastritis (CAG), Helicobacter pylori gastritis, celiac disease, and inflammatory bowel disease (IBD). It also complicates the use of gastric acid–suppressing therapies, which leads to chronic hypergastrinemia in most patients (58).
various chronic inflammatory conditions, including chronic atrophic gastritis (CAG), Helicobacter pylori gastritis, celiac disease, and inflammatory bowel disease (IBD). It also complicates the use of gastric acid–suppressing therapies, which leads to chronic hypergastrinemia in most patients (58).
TABLE 17.3 Proliferative Endocrine Cell Lesion | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
NE cell hyperplasia results from a combination of a prolonged cell half-life, augmented replication of mature cells, and/or differentiation of a larger fraction of uncommitted stem cells into specific NE cell types. Each of these mechanisms may be triggered by loss of normal inhibitory influences on NE cell proliferation, a lack of normal negative feedback mechanisms, or the trophic action of stimulating substances. Additionally, autocrine mechanisms may stimulate a cell’s own proliferation. The local tissue microenvironment may also play a significant role in inducing the hyperplasia.
The stages in the progression of hyperplastic neuroendocrine lesions are shown in Table 17.3. Simple or diffuse hyperplasia, the earliest hyperplastic stage, is characterized by a diffuse increase in NE cells scattered singly or in clusters of up to three cells per gland. The cells may appear enlarged, especially in the lower third of the mucosa. Linear hyperplasia is diagnosed when linear, semi-linear, or daisy-chain–like ECL cell configurations are present. The next stage, micronodular hyperplasia, consists of solid micronodular NE cell nests measuring 100 to 150 μm in size (the average diameter of a gastric gland) (57,59,60). These may be bounded by an intact basement membrane continuous with that of the rest of the gland or lie in the basal glandular portion of the mucosa dissociated from the glands lying free in the lamina propria abutting the muscular mucosae. Adenomatoid hyperplasia consists of a close aggregation of five or more interglandular micronodular lesions, each with an intact basement membrane. As each micronodule enlarges, it breaks down its basement membrane and the cells develop cytologic atypia with an increased nuclear:cytoplasmic ratio, thereby qualifying for a diagnosis of a dysplasia. This dysplastic stage marks the borderline between the hyperplastic stages preceding it and the neoplastic stage of a fully developed carcinoid tumor following it. It is the earliest “point of no return” in the hyperplasia–neoplasia sequence of ECL proliferation (57). These lesions include enlarging micronodules, fusing micronodules, microinvasive lesions, and nodules with newly formed stroma. The carcinoid stage is characterized by nodular infiltrating growths measuring >0.5 mm in diameter. These steps may be seen in proliferative NE cell lesions involving many cell types including G, ECL, EC, D, and L cells.
Often, gastrointestinal NE cell hyperplasia is unsuspected (61), since most lesions do not release enough hormones or other secretory products to produce significant biochemical abnormalities or to give rise to specific clinical syndromes. Furthermore, even though hyperplasia may be present, the lesions may not be visible endoscopically or grossly. Additionally, NE cell hyperplasias often remain unrecognized even when standard H&E-stained sections are examined due to their rarity and the difficulty in recognizing increased numbers of endocrine cells among the other epithelial cells present in the glands. Sometimes, it is only when suspicions of the gastroenterologist and/or pathologist cause the specimen to be stained a neuroendocrine marker that the increased number of endocrine cells becomes apparent.
Neuroendocrine Cell Hyperplasia in the Esophagus
NE cell hyperplasia in the esophagus usually develops in the setting of Barrett esophagus (BE). It is a common event affecting up to 90% of cases of BE. The majority of the cells are EC cells (62).
Neuroendocrine Cell Hyperplasia in the Stomach
G-cell Hyperplasia
G cells become hyperplastic in any condition that lowers gastric acid concentrations, including extensive multifocal gastritis,
pernicious anemia, vagotomy, and prolonged treatment with proton pump inhibitors (Table 17.4; Figs. 17.12, 17.13, and 17.14). G-cell hyperplasia following vagotomy results from a combination of chronic antral distention, decreased acid secretion, and release from vagally mediated inhibitors of G-cell prolifera-tion. G-cell hyperplasia in acromegaly results either from the patients also having the MEN-1 syndrome or from unidentified trophic factors secreted by the pituitary.
pernicious anemia, vagotomy, and prolonged treatment with proton pump inhibitors (Table 17.4; Figs. 17.12, 17.13, and 17.14). G-cell hyperplasia following vagotomy results from a combination of chronic antral distention, decreased acid secretion, and release from vagally mediated inhibitors of G-cell prolifera-tion. G-cell hyperplasia in acromegaly results either from the patients also having the MEN-1 syndrome or from unidentified trophic factors secreted by the pituitary.
TABLE 17.4 Conditions Associated with Hypergastrinemia and G-cell Hyperplasia | |
|---|---|
|
Primary antral G-cell hyperplasia and hyperfunction is a rare pediatric disorder in which G-cell hyperplasia, hyperfunction, or both occur without a clear cause (63). This entity, also known as pseudo-Zollinger-Ellison syndrome, causes clinical and biochemical features resembling those seen in Zollinger-Ellison syndrome (ZES) but the patients lack a gastrin-producing tumor. The hypergastrinemia reverts to normal following antrectomy. Most familial cases result from a genetic defect in normal regulation of G-cell function or proliferation. Patients with nonfamilial forms of the disease have increased antral G-cell sensitivity to intragastric food stimulation (64).
The distribution of hyperplastic G-cell populations in primary and secondary antral G-cell hyperplasia is similar with increased G-cell numbers in the lower and middle thirds of the antral glands. Occasionally, longstanding diffuse antral G-cell hyperplasia progresses to multiple small micronodular G-cell clusters (Fig. 17.14) that eventually give rise to G-cell tumors (gastrinomas). Gastrin also stimulates oxyntic mucosal growth and increases the progenitor cell labeling index in this area.
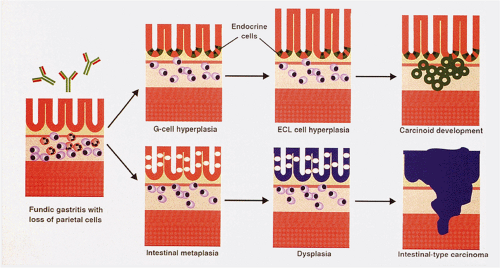 FIG. 17.12. Progression of autoimmune gastritis to carcinoid tumors and intestinal-type carcinomas. Patients with autoimmune gastritis develop G-cell hyperplasia in response to an antibody-mediated attack on the parietal cells that subsequently leads to decreased acid production. The hyperplastic G cells produce increased amounts of gastrin, leading to both foveolar hyperplasia as well as ECL cell hyperplasia. The ECL cell hyperplasia progresses further to the development of ECL micronests and eventually carcinoid tumors. At the same time, the gastric epithelium may become intestinalized in response to epithelial loss from the immunologic attack. This intestinal epithelium becomes dysplastic and can progress to intestinal-type carcinomas. |
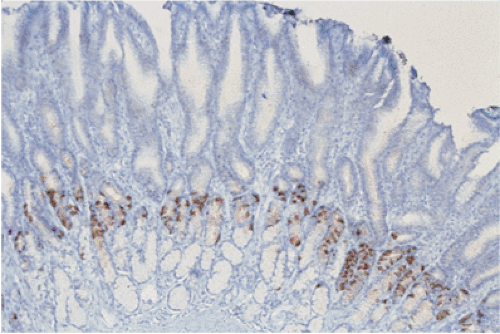 FIG. 17.13. Linear G-cell hyperplasia highlighted by gastrin immunostain. |
Enterochromaffinlike Cell Hyperplasia
ECL cells are extremely sensitive to the stimulatory effects of gastrin, undergoing secondary hyperplasia in chronic hypergastrinemic states (65). Hypergastrinemia results in increased proliferation of both pluripotential stem cells and mature ECL cells (66). Gastrin also stimulates ECL function, as reflected by an increase in histamine decarboxylase activity and histamine release (67). The ECL hyperplasia is reversible if the hypergastrinemia is removed. ECL lesions develop exclusively in the oxyntic mucosa. In CAG, the hyperplastic cells are present only in the atrophic fundic glands and pyloric metaplastic glands but not in the intestinal metaplastic glands. Hyperplastic ECL lesions range from a diffuse ECL cell hyperplasia through linear and nodular aggregates to intramucosal and frankly invasive carcinoid tumors (Figs. 17.15 and 17.16). Patients with hypergastrinemia due to H. pylori infections or the chronic use of proton pump inhibitors (68) develop ECL cell hyperplasia, but carcinoid tumors are rare.
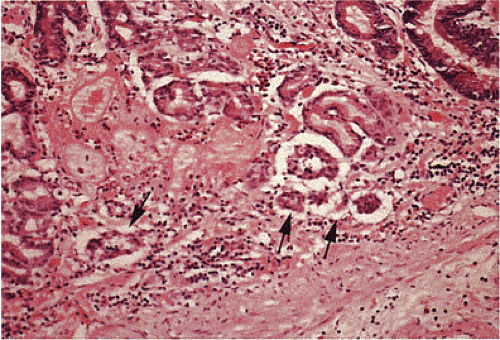 FIG. 17.14. Antral G-cell hyperplasia in a patient with autoimmune gastritis. Note the clusters of endocrine cells in the lamina propria (arrows). The lesion is visible even without a gastrin or neuroendocrine immunostain. The hyperplastic focus corresponds to an area of micronodular hyperplasia. |
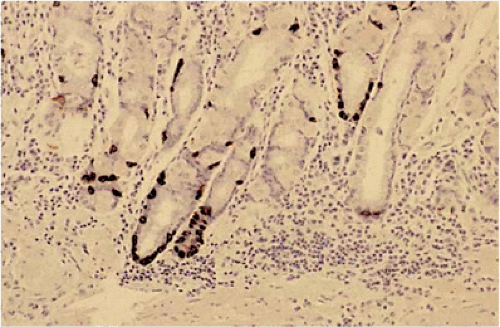 FIG. 17.15. Chromogranin immunostain showing linear hyperplasia. |
The most frequent histologic change in ECL cells in ZES patients is a diffuse hyperplasia, affecting more than half of ZES patients. Linear hyperplasia affects approximately 18% of patients, whereas micronodular hyperplasia is infrequent (59). Dysplastic ECL lesions can also develop. The topographic distribution of linear hyperplasia often localizes to the greater curvature and does not correspond to that of the ECL cell micronodular hyperplasia, dysplasia, and ECL tumor, changes that are more uniformly distributed between the lesser and greater curvatures. This observation led Bordi to suggest that the linear hyperplasia occurring in the setting of ZES may not be a step in the micronodular hyperplasia–dysplasia–NET (carcinoid) sequence. Rather, linear hyperplasia may represent a self-limited lesion (59). Antrectomy as well as some pharmacologic treatments may reverse the ECL cell hyperplasia.
D-cell Hyperplasia
Antral D-cell hyperplasia develops in patients with duodenal ulcer disease (69). Extensive gastric D-cell hyperplasia was reported in a 37-year-old woman with dwarfism, obesity, dryness of the mouth, and goiter. The D-cell density in this patient was increased 39-fold in the gastric fundus and 25-fold in the antrum. Antral G cells were increased 2.3-fold and they showed pronounced hypertrophy (69). The hypersomatostatinemia may have interfered with pituitary hormone release at an early age, thereby resulting in the dwarfism.
Hyperplasia of Ghrelin-producing Cells
Pericarcinoidal oxyntic mucosal ghrelin cell hyperplasia consistently occurs in patients with CAG. The ghrelin-producing cells are larger than their normal counterparts and appear polygonal or flask shaped. Diffuse, linear, and nodular forms of hyperplasia occur (19).
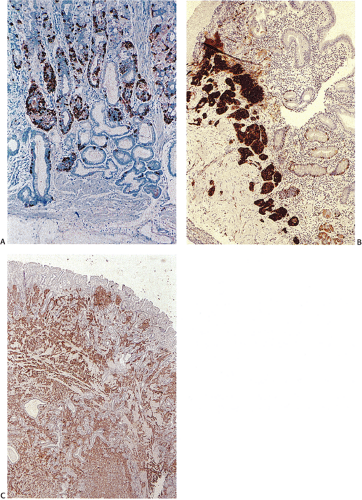 FIG. 17.16. Enterochromaffinlike (ECL) cell hyperplasia. A: Linear hyperplasia with micronests. B: Dysplastic ECL cell nodules with newly formed stroma. C: Fully developed ECL cell carcinoid tumor infiltrating the mucosa and submucosa. Chromogranin immunostain. |
Neuroendocrine Cell Hyperplasia in the Intestines
EC cell hyperplasia occurs in the intestines in three major settings: Celiac disease, inflammatory bowel disease, and adjacent to carcinoid tumors. D-cell hyperplasia may be present in patients with somatostatinomas and MEN-1. In the setting of celiac disease, hyperplastic NE cells are irregularly distributed within the crypts, often associated with increased numbers of Paneth cells. Goblet cells are normal or occasionally increased in number. The subepithelial basement
membrane may appear normal or thickened (Fig. 17.17) (70). These changes progress with increased mucosal flattening and expansion of the crypt cells. Pyloric metaplasia may develop. The abnormalities are most marked on the crests of mucosal folds.
membrane may appear normal or thickened (Fig. 17.17) (70). These changes progress with increased mucosal flattening and expansion of the crypt cells. Pyloric metaplasia may develop. The abnormalities are most marked on the crests of mucosal folds.
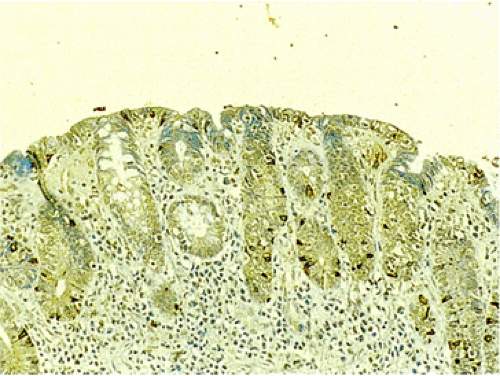 FIG. 17.17. Endocrine cell hyperplasia in the completely flattened small intestinal mucosa of a patient with celiac disease. Chromogranin immunostain. |
TABLE 17.5 World Health Organization Classification of Endocrine Tumors | |
|---|---|
|
Endocrine cell hyperplasia affects as many as 40% of IBD patients (71). Paneth cell metaplasia distal to the hepatic flexure, pyloric metaplasia, and endocrine cell hyperplasia often indicate a long history of colitis (72). IBD patients may also develop very small NE tumors, called microcarcinoids. They are not grossly evident and are typically detected in biopsy specimens performed for other reasons or in resection specimens. When present, these lesions lie in the muscularis mucosae and upper submucosa (73). The cells are arranged in a trabecular fashion and consist of multiple small islands of large, pale, eosinophilic cells.
Endocrine cell micronests (ECMs) occur in the minor and major duodenal papillae and are usually immunoreactive for somatostatin and pancreatic polypeptide (74). Some of the lesions break off the ductules of the pancreatic duct or represent atrophic or degenerating islets. Some patients exhibit rectal endocrine cell hyperplasia or focal NE micronests, often surrounding rectal carcinoid tumors (Fig. 17.18) (75). Microcarcinoids may develop in areas of diversion colitis (76).
Terminology of Neuroendocrine Tumors
NE tumors range from classic carcinoid tumors to small cell carcinomas. They may be classified according to their location, the normal cell counterpart toward which differentiation is occurring, and whether they are benign, of borderline malignancy, of low-grade malignancy, or of high-grade malignancy (77,78). Tumors of mixed endocrine and glandular lineage are classified separately. Carcinoid tumors are classically defined as low-grade, potentially malignant, epithelial neoplasms showing NE differentiation. They most frequently develop in the gastrointestinal system. The term carcinoid tumor, as commonly used by pathologists, encompasses a wide spectrum of neoplasms that originate from various NE cells. However, it has become clear that not all gastrointestinal carcinoid tumors are the same. Rather, they reflect the products they secrete and the cells from which they arise. Thus, gastric carcinoids differ substantially from ileal carcinoids, which differ significantly from appendiceal lesions. Current estimates indicate that the various gastrointestinal carcinoid tumors produce as many as 40 different secretory products (79). These tumors may be referred to by the name of the cell population from which they arise (such as ECL tumors), by the hormones they produce (such as gastrinoma), or by their location in the gut (such as midgut carcinoid). It has recently been suggested that these tumors be called well-differentiated neuroendocrine tumors (78), terminology adopted by the latest World Health Organization (WHO) classification (Table 17.5) (77). However, to avoid confusion, the term carcinoid was not entirely abandoned in the revised classification.
Well-Differentiated Neuroendocrine Tumors (Carcinoid Tumors)
Fifty-four to eighty-five percent of all carcinoid tumors arise in the GI tract (80). They develop anywhere, from the esophagus to the anus, but they are decidedly unusual in the esophagus and anus. The widespread use of endoscopy, ultrasonography, computerized tomography, magnetic resonance imaging, and other imaging techniques have significantly enhanced the detection of previously undetectable lesions. For this reason, there appears to have been an increase in the incidence of gastrointestinal carcinoid tumors as well as a shift in the relative proportions of tumors arising in various sites (81). In a recent population-based cancer registry study, the gastrointestinal tract accounted for 54% of NETs. Within the gut, the small intestine was the most common site (44.7%), followed by the rectum (19.6%), appendix (16.7%), colon (10.6%), and stomach (7.2%) (81).
Some NETs develop in unusual locations, such as in Meckel diverticula (Fig. 17.19) (82), cystic duplications (83), and the mesentery (84). These neoplasms often arise on a background of NE cell hyperplasia.
Some NETs develop in unusual locations, such as in Meckel diverticula (Fig. 17.19) (82), cystic duplications (83), and the mesentery (84). These neoplasms often arise on a background of NE cell hyperplasia.
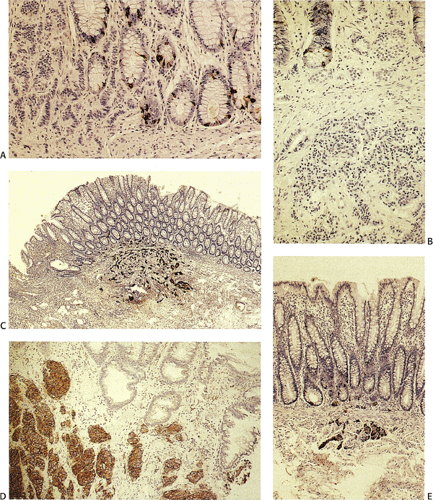 FIG. 17.18. Rectal endocrine cell hyperplasia and carcinoid tumors. A: Endocrine cell hyperplasia adjacent to a carcinoid tumor (chromogranin immunostain). Note that the carcinoid tumor (left) is chromogranin negative. B: A carcinoid tumor lies in the superficial submucosa. The overlying rectal mucosa shows endocrine cell hyperplasia. C: Small rectal carcinoid found incidentally at the time of histologic examination. D: Synaptophysin-stained adenocarcinoid tumor. E: Microcarcinoid straddling the muscularis mucosae. This was one of multiple small carcinoid tumors present in the rectum of this patient. There is overlying endocrine hyperplasia in the colonic mucosa. |
Foregut NETs include esophageal, gastric, and proximal duodenal tumors. Midgut lesions include distal duodenal, small intestinal, ascending colon, and proximal transverse colon tumors. Hindgut NETs include distal transverse colon, descending colon, and rectal tumors. While this concept is probably outdated today, it has historical value since tumors derived from these embryologic origins differ clinically, histochemically, and immunohistochemically. Foregut and hindgut tumors are typically argentaffin negative in contrast with midgut tumors, which are argentaffinic.
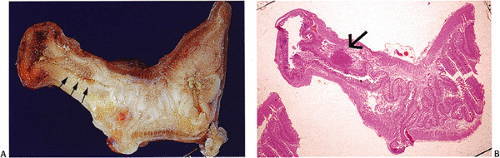 FIG. 17.19. Incidental carcinoid arising in Meckel diverticulum. A: The lesion is not readily visible in the gross specimen unless one looks carefully for it (arrows). B: The tumor (arrow) is well demarcated but unencapsulated. |
NETs are usually easily diagnosed because of their distinctive histologic appearance. They exhibit five histologic patterns (Table 17.6) (85). The tumors contain cytoplasmic secretory granules identifiable by the techniques previously discussed. A tumor should only be diagnosed as a carcinoid tumor if it displays the classic histologic architecture of trabecular, insular, or ribbonlike cell clusters with little or no cellular pleomorphism and sparse mitoses. The presence of focal endocrine differentiation in a tumor lacking classic histologic features should not be diagnosed as a NET.
The clinical behavior of this group of tumors is often unpredictable and the traditional morphologic criteria of malignancy have limited applicability to NETs. Tumor biology is influenced by tumor size, location, stage, evidence of metastasis, and histologic features (78). Thus, tumors arising in the appendix are often found incidentally and are usually small and seldom metastasize (86). In contrast, ileal and jejunal tumors are frequently already metastatic to the regional lymph nodes and liver when they are first diagnosed (87). Patient prognosis may also be influenced by the clinical setting in which the tumors arise, the presence or absence of the carcinoid syndrome, and the underlying molecular abnormalities. These features are discussed further in the context of the site-specific NETs.
Some NETs may present early when there is an overt syndrome such as that related to a gastrinoma, but other tumors present at a late stage with liver metastases and the carcinoid syndrome and metastatic tumor in the liver. The measurement of tumor markers in the circulation of patients with NETs may be useful and is of threefold importance. It often establishes the diagnosis, it is useful in monitoring disease progression and response to treatment, and it may serve as a prognostic marker. For example, circulating CgA is elevated in approximately 90% of malignant gastrointestinal NETs (88). In addition, many neoplastic NE cell proliferations express somatostatin receptors (SSTRs), allowing new diagnostic and therapeutic strategies to be developed. Somatostatin analogs reduce secretion via inhibitory G proteins and therefore may lead to marked symptom relief. By using radiolabeled somatostatin analogs, tumors can be localized scintigraphically or during surgery with the aid of scintillation detection (radioguided surgery). Residual tumors may be treated with radionuclide therapy since radionuclides are internalized into tumor cells after SSTR binding (89).
TABLE 17.6 Histologic Patterns of Carcinoid Tumors | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
One problem that arises in the diagnosis of NETs is distinguishing a gastrointestinal carcinoid tumor from a pulmonary carcinoid tumor. The panel of cytokeratin (CK)-20/CK-7 and thyroid transcription factor (TTF)-1 antibodies may distinguish among these lesions, since pulmonary tumors do not express CK-20 and GI tumors usually do not express TTF-1 or CK-7 (90).
Esophageal Carcinoid Tumors
Esophageal NETs are very rare, accounting for 0.05% of all gastrointestinal carcinoids and 0.02% of all esophageal carcinomas. The majority of patients are males with an age range of 30 to 82 and a mean age of 56 to 63 years (91,92). The tumors usually develop in the lower esophagus, are typically solitary lesions, and may arise in association with adenocarcinoma in the setting of Barrett esophagus (92). Patients present with dysphagia. The development of the carcinoid syndrome is extremely rare (93). The tumors tend to be large, measuring up to 12 cm in diameter, and they
may be restricted to the lamina propria or deeply infiltrate the esophageal wall (92,94,95). Some tumors appear polypoid. Metastases are present in approximately 50% of cases (95). Histologically, foregut NETs contain anastomosing ribbons, solid nests, trabeculae, acini, or rosettes. The cells are usually round or polygonal but they may appear oval or cylindrical and may have a high mitotic rate (4 mitoses/10 high-powered field [hpf]) (94,95). Carcinoid tumors that arise in the setting of BE-associated adenocarcinoma exhibit endocrine cell hyperplasia in the associated adenocarcinoma. The tumors are reported to have a poor prognosis even in the absence of metastases (95), but this issue may be confused by the inclusion of atypical carcinoids or even small cell carcinomas in some reports of these lesions (92). Long-term survivors are reported (96) even with nodal metastases.
may be restricted to the lamina propria or deeply infiltrate the esophageal wall (92,94,95). Some tumors appear polypoid. Metastases are present in approximately 50% of cases (95). Histologically, foregut NETs contain anastomosing ribbons, solid nests, trabeculae, acini, or rosettes. The cells are usually round or polygonal but they may appear oval or cylindrical and may have a high mitotic rate (4 mitoses/10 high-powered field [hpf]) (94,95). Carcinoid tumors that arise in the setting of BE-associated adenocarcinoma exhibit endocrine cell hyperplasia in the associated adenocarcinoma. The tumors are reported to have a poor prognosis even in the absence of metastases (95), but this issue may be confused by the inclusion of atypical carcinoids or even small cell carcinomas in some reports of these lesions (92). Long-term survivors are reported (96) even with nodal metastases.
TABLE 17.7 Types of Gastric Carcinoid Tumors | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||
TABLE 17.8 Classification of Neuroendocrine Tumors of the Stomach | |||||||
|---|---|---|---|---|---|---|---|
|
Gastric Carcinoid Tumors
Gastric NETs arise from endocrine cells of the gastric mucosa, usually the ECL cells. They develop in five settings, four of which associate with ECL cell hyperplasia (Table 17.7). ECL-type tumors are frequently classified according to the Rindi classification that recognizes the first three types of tumors listed in Table 17.7 (97). Table 17.8 shows the latest WHO classification of gastric endocrine cell tumors (78). Tumors arising in the various settings differ significantly in their biologic behavior (98,99). In a large series of ECL carcinoids, 79% were associated with CAG, 10% of patients had ZES, and 11% of tumors were sporadic (97).
In the past, gastric NETs accounted for about 4% of the total number of gastrointestinal carcinoids (100) and 0.3% of gastric neoplasms (141). However, these lesions have become more common, related to improved diagnostic methods including endoscopy and immunohistochemistry and heightened awareness of their existence. They are now estimated to account for 11% to 30% of GI NETs (101). In Japan, a country with extensive use of gastric endoscopy, the stomach is the second most common site of NETs. The increased use of acid-suppressing therapies may also account for an increasing incidence of ECL cell proliferative lesions. Up to 100% of patients on high-dose acid suppressive therapies develop hypergastrinemia (58) and the possibility of developing NETs.
ECL cell proliferations (diffuse, linear, and/or micronodular hyperplasias) and their more advanced equivalents (ECL cell dysplasia and ECL carcinoid tumors) all associate with chronic hypergastrinemia (102). However, hypergastrinemia alone appears to be insufficient to induce carcinoid
development as evidenced by the fact that ZES patients without the MEN-1 syndrome rarely develop gastric carcinoids despite prolonged serum gastrin levels more than 10-fold normal (99). In contrast, patients with familial MEN-1 who develop ZES, develop type II carcinoids 30 times more frequently than the normal population. These carcinoid tumors affect 13% to 30% of all MEN ZES cases (103). Therefore, both endocrine and genetic factors are implicated in the pathogenesis of type I and type II gastric carcinoids.
development as evidenced by the fact that ZES patients without the MEN-1 syndrome rarely develop gastric carcinoids despite prolonged serum gastrin levels more than 10-fold normal (99). In contrast, patients with familial MEN-1 who develop ZES, develop type II carcinoids 30 times more frequently than the normal population. These carcinoid tumors affect 13% to 30% of all MEN ZES cases (103). Therefore, both endocrine and genetic factors are implicated in the pathogenesis of type I and type II gastric carcinoids.
Genetic traits, as in patients with MEN-1; other genetic alterations, as discussed earlier; overexpression of the gastrin receptor gene or growth factors such as EGF or TGF-α; and alterations in the local microenvironment may all play a role in the subsequent development of the carcinoid tumors. Other substances may also play a role in tumor development. Recently it has been shown that pituitary adenylate cyclase–activating polypeptide and vasoactive intestinal polypeptide are even more potent modulators of ECL hyperplasia than gastrin (102). The reg-protein may also control ECL cell numbers by restraining the stimulatory effect of gastrin.
Neuroendocrine Tumors Arising in the Setting of Chronic Atrophic Gastritis (Type I Tumors)
Type I carcinoids account for approximately 74% to 79% of all gastric NETs. Five to ten percent of patients with CAG and hypergastrinemia develop ECL carcinoids (98,102). The demographics of the lesion mimic those of autoimmune gastritis. More than 70% of cases develop in older women with a mean age of 63 years (98,99). Type I carcinoids develop in part from the hypergastrinemia that results from loss of parietal cell mass and decreased acid output as a consequence of the autoimmune gastritis (98,99). Tumors developing in this setting arise on a background of ECL cell hyperplasia and dysplasia (Fig. 17.20). In more than half of the cases, the tumors are multifocal.
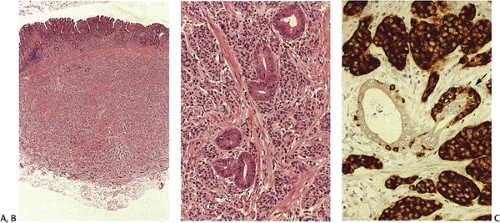 FIG. 17.20. Carcinoid tumor. A: Low-power view demonstrates a proliferation of small, uniform cells within the deep mucosa and submucosa. B: The cells are arranged in nests and rosettelike structures. There is no nuclear pleomorphism and mitotic figures are not seen. Residual gastric glands lie among the tumor cells. C: The endocrine differentiation of this lesion is highlighted by the chromogranin stain. A focus of linear endocrine hyperplasia is present in a residual gastric gland (arrow). |
Type I carcinoids are the most “benign” of the gastric carcinoid tumors (104). They present as small, tan submucosal nodules or polyps in the corpus usually measuring <1 cm. Most measure only 1 to 3 mm in diameter; 97% are <1.5 cm in diameter. The tumors tend to be limited to the mucosa and submucosa, with only 7% invading the muscularis propria. They infrequently cause liver metastases (2% to 5% of cases) (98).
Most tumors are characterized by small microlobular aggregates formed by regularly distributed cells that may create a mosaiclike pattern. They consist of solid nests, trabeculae, ribbons, tubular structures, or a combination of these patterns. The cytoplasm appears slightly eosinophilic, slightly basophilic, or amphophilic. Cytoplasmic granules may or may not be seen on routine preparations. The cells contain centrally located, round or oval, monomorphic nuclei, with generally inapparent nucleoli and little or no nuclear pleomorphism or mitotic activity. Angioinvasion is rare. The background mucosa shows the typical features of CAG, often with intestinal metaplasia.
The tumor cells are argyrophilic but not argentaffinic. They are consistently immunoreactive for NE markers including CgA, synaptophysin, and NSE. The tumor cells, as well as the surrounding hyperplastic endocrine cells, are positive for the newly discovered hormone ghrelin (19). VMAT-2 is a specific marker for ECL tumors. The cells variably produce mucin (105). A small population of tumor cells
may be positive for serotonin, gastrin, somatostatin, pancreatic polypeptide, or α-human chorionic gonadotropin (α-hCG). A few ECL cell tumors produce histamine and 5-hydroxytryptophan (5-HT); when these lesions metastasize they may produce an atypical carcinoid syndrome (97).
may be positive for serotonin, gastrin, somatostatin, pancreatic polypeptide, or α-human chorionic gonadotropin (α-hCG). A few ECL cell tumors produce histamine and 5-hydroxytryptophan (5-HT); when these lesions metastasize they may produce an atypical carcinoid syndrome (97).
Type I carcinoid tumors may regress after gastrin levels decrease, usually by antrectomy (106) or treatment with somatostatin analogs (107). These treatments result in resolution, regression, or stabilization of many ECL tumors (108). At some stage, however, ECL cell proliferations may become irreversible, making it difficult to estimate the degree of tumor regression following treatment. It is hard to predict which of multiple ECL tumors have progressed beyond the point of gastrin dependence and are autonomous in their growth. The octreotide suppression test may predict a beneficial outcome from antrectomy (109). The tumors may also be removed surgically or endoscopically depending on their size and number (110). Biopsy and observation are other possible therapeutic options. Gastrectomy is reserved for patients with extensive tumor involvement of the gastric wall, to control bleeding, or in patients in whom the tumors progress (110).
When the tumors metastasize, it is usually to the regional lymph nodes; distant metastases are rare. They almost never cause the death of the patient (98). The generally better prognosis of this type of gastric NET relates in part to earlier diagnosis because the patients are commonly seen for symptoms related to the underlying CAG. Alternatively, the better prognosis may relate to the intrinsically benign nature of tumors arising in this setting.
Neuroendocrine Tumors Arising in the Setting of Multiple Endocrine Neoplasia Type 1–Zollinger-Ellison Syndrome (Type II Carcinoid Tumors)
Type II carcinoids represent 6% to 10% of all gastric NETs. The tumors do not show any gender predilection and occur at an earlier age than type I tumors with a median age at development of 50 years (98). Gastric carcinoids arising in the setting of ZES usually associate with MEN-1 (98). The ECL cell NETs develop in the oxyntic mucosa and are frequently multiple (136). They are the consequence of hypergastrinemia resulting from a gastrin-secreting NE cell neoplasm (often a pancreatic islet cell tumor). Rare sporadic ZES-associated gastric carcinoids also occur, although generally these patients only have simple linear hyperplasia (57).
ECL NETs arise on a background of parietal cell hypertrophy and hyperplasia and ECL cell hyperplasia and dysplasia. Since the patients present with a hypertrophic gastropathy characteristic of ZES, the gastric folds appear thickened. In addition, there are usually multiple, small tumor nodules. ECL tumors occurring in this setting are more likely to measure >1.5 cm and to produce local lymph node metastases (30%) and liver metastases (10%) than type I tumors (98). The histologic features of the tumors resemble type I gastric carcinoids described above.
Neuroendocrine Tumors Arising in the Setting of Achlorhydria and Parietal Cell Hyperplasia
Recently two patients were reported with multiple gastric carcinoids arising in the setting of marked hypergastrinemia, associated with achlorhydria and hypertrophic parietal cells. The parietal cell hyperplasia caused mucosal thickening. Focal intestinal metaplasia was present. There were also areas of ciliated metaplasia. Multiple intramucosal and invasive carcinoid tumors involving the gastric body and fundus arose on a background of marked ECL cell hyperplasia. The largest tumor nodule measured 1.3 cm in diameter. A micrometastasis was present in a regional lymph node. The oxyntic mucosa showed marked parietal cell hyperplasia and hypertrophy. Some of the parietal cells appeared vacuolated and many displayed protrusions of their apical cytoplasm into dilated oxyntic glands filled with inspissated eosinophilic material. (113,114). This presentation appears to represent a rare form of gastric carcinoids associated with an intrinsic acid secretion abnormality as supported by the ultrastructural demonstration of the lack of the parietal cell canalicular system (114) and the failure of the parietal cells to stain with an antibody directed at the α and β subunits of the H+/K+-ATPase proton pump (113).
Sporadic Gastric Neuroendocrine Tumors (Type III Carcinoids) (Well-differentiated Neuroendocrine Carcinomas)
Sporadic NETs are most common in men (74%), with a mean age of 55 years (98). They are unassociated with CAG and hypergastrinemia and represent a proliferation of various cell types including ECL, EC, and X cells. They are usually single lesions that do not arise on a background of NE cell hyperplasia. The incidence of multiple type III carcinoids is only 1% (98). The tumors occur anywhere in the stomach. Unlike type I and type II tumors, type III tumors are aggressive and larger than other gastric NETs. They generally present as a mass lesion mimicking the clinical presentation of adenocarcinomas with bleeding, obstruction, or metastasis as presenting findings. Patients may present with an atypical carcinoid syndrome with cutaneous flushing in the absence of diarrhea. Tumors producing these clinical symptoms usually produce histamine and 5-HT (115).
Grossly, type III tumors often appear as smooth, round, yellow, submucosal nodules measuring >2 cm in diameter (Fig. 17.21). They are usually covered by an intact mucosa, although larger lesions may develop an irregularly shaped, reddened depression or surface ulceration. Histologically, type III NETs exhibit various growth patterns, including trabecular, gyriform, medullary or solid, glandular, or rosette, or a mixture of all of these (Fig. 17.22). Often they show a
prevalence of solid cellular aggregates (Fig 17.23) and large trabeculae with cellular crowding. The round, spindle-shaped, and polyhedral cells are irregularly distributed and have fairly large vesicular nuclei and prominent small, eosinophilic nucleoli. The cells may also contain smaller hyperchromatic nuclei with irregular chromatin clumping and increased mitoses. Sparse areas of necrosis may be present. Larger tumors may invade deeply and promote a prominent fibroblastic stromal response. These lesions extend intramurally and invade vascular or lymphatic channels to produce nodal and distant metastases (116). Factors that predict aggressive behavior include cellular atypia, two or more mitoses per hpf, angioinvasion, and transmural invasion (98,117). Although tumor size correlates with a
tendency to metastasize, minute tumors are known to spread beyond the stomach (118). Thus, while the latest WHO criteria suggest that NETs tumors ≤1 cm are benign lesions, this criterion does not appear to be applicable to all of these tumors. Regional lymph node metastasis may be seen in tumors as small as 0.3 cm in diameter (119).
prevalence of solid cellular aggregates (Fig 17.23) and large trabeculae with cellular crowding. The round, spindle-shaped, and polyhedral cells are irregularly distributed and have fairly large vesicular nuclei and prominent small, eosinophilic nucleoli. The cells may also contain smaller hyperchromatic nuclei with irregular chromatin clumping and increased mitoses. Sparse areas of necrosis may be present. Larger tumors may invade deeply and promote a prominent fibroblastic stromal response. These lesions extend intramurally and invade vascular or lymphatic channels to produce nodal and distant metastases (116). Factors that predict aggressive behavior include cellular atypia, two or more mitoses per hpf, angioinvasion, and transmural invasion (98,117). Although tumor size correlates with a
tendency to metastasize, minute tumors are known to spread beyond the stomach (118). Thus, while the latest WHO criteria suggest that NETs tumors ≤1 cm are benign lesions, this criterion does not appear to be applicable to all of these tumors. Regional lymph node metastasis may be seen in tumors as small as 0.3 cm in diameter (119).
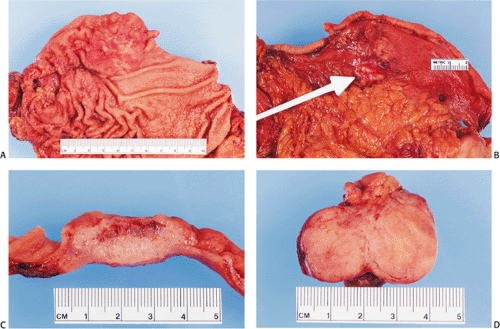 FIG. 17.21. Gross appearance of sporadic gastric carcinoid tumor. A: Opened stomach showing the tumor. B: The tumor extends to the serosal surface (arrow). C: Cut surface of the tumor showing that it extends through the gastric wall. D: Metastatic tumor in a perigastric lymph node. |
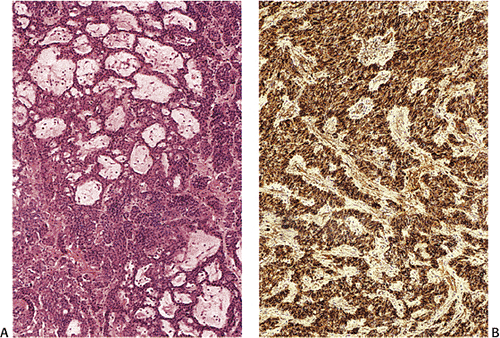 FIG. 17.22. Gastric carcinoid tumor. A: The tumor is composed of solid nests, ribbons, and glandlike structures. The cells are uniform and mucin has accumulated in many areas. B: Chromogranin stain highlights the cords of cells. |
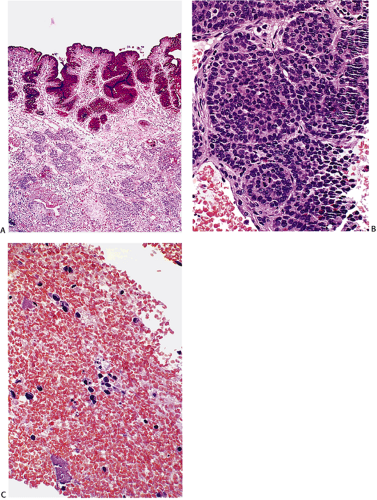 FIG. 17.23. Gastric carcinoid. A: The tumor is completely submucosal in location. Alcian blue periodic acid–Schiff (PAS) stain. The neutral mucins of the gastric epithelium are strongly PAS positive. The carcinoid tumor is negative. B: The tumor nests are solid with peripheral palisading. C: Bone marrow metastases. |
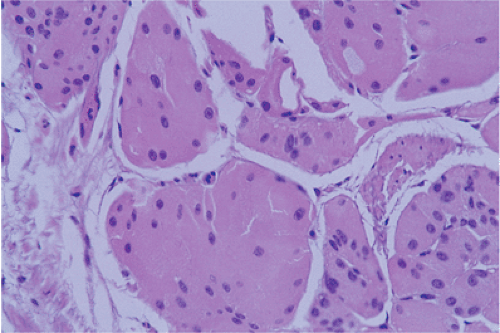 FIG. 17.24. Gastric carcinoid with oncocytic features. |
These tumors frequently express p53 and have a high KI-67 labeling rate. Rare tumors show widespread ossification or have an oncocytic appearance (Fig. 17.24). These tumors are also positive for markers of bone formation and differentiation including bone morphogenetic protein, osteopontin, and osteonectin (120). Type III carcinoid tumors often display an aggressive local behavior and metastasize. Seventy-six percent of tumors invade the muscularis propria and/or subserosa and lymph node, and distant metastases are present in 2% to 71% and 22% to 75% of cases, respectively (98). The 5-year survival in patients with sporadic carcinoids is <50%, reflecting the high rate of metastasis. The 5-year survival rate is significantly higher for localized disease (64.3%) and for those with regional metastases (29.9%) than for lesions with distant metastases (10%) (91). As noted above, patients with type I carcinoid tumors benefit from antrectomy, but this treatment has no value in sporadic carcinoid tumors (121). Therefore, it is important to distinguish between these two groups of patients. This is usually accomplished based on clinical and morphologic features, as well as on the presence or absence of adjacent ECL cell hyperplasia. The features of sporadic and hypergastrinemia-associated carcinoids are compared in Table 17.9.
Non–enterochromaffinlike Neuroendocrine Tumors
These tumors arise anywhere in the stomach and are usually single large lesions that are often highly malignant. Affected patients may present with ZES due to gastrin production by the tumor or with Cushing syndrome due to secretion of adrenocorticotropic hormone (ACTH). Serotonin-producing NETs are rare in the stomach (98). They resemble midgut carcinoids, consisting of rounded nests of tightly packed small tumor cells often showing peripheral palisading. Gastrin-producing tumors exhibit a characteristic thin trabecular pattern and the cells are strongly positive for NE markers and for gastrin. These tumors may metastasize and also cause ZES.
TABLE 17.9 Comparison of Sporadic and Hypergastrinemia-associated Carcinoid Tumors | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Small Intestinal Neuroendocrine Tumors
Duodenal Neuroendocrine Tumors
It used to be believed that duodenal NETs only accounted for 1.8% to 2.9% of gastrointestinal NETs (100), but with the advent of improved imaging and the increased use of upper endoscopy, they are currently estimated to account for up to 22% of all gut endocrine tumors (80). Gastrin cell tumors account for the largest group (62% to 65%) of tumors arising in the upper intestine, followed by somatostatin cell tumors (15% to 21%), gangliocytic paragangliomas (9%), undifferentiated tumors, and pancreatic polypeptide (PP) cell tumors (122). The WHO classification of these tumors is shown in Table 17.10. In one study, 72% of patients with duodenal NETs were male. Patient median age was 53 to 59 years with a range of 18 to 90 years (123). These tumors associate with MEN-1, ZES, and NF1. The majority of duodenal carcinoids exhibit a mixture of cribriform, insular, glandular, and solid and trabecular growth patterns. They produce various hormones as discussed below. In addition, xenin is said
to be a marker of duodenal NETs because it is exclusively expressed in these tumors, regardless of their hormonal content and functional activity (124).
to be a marker of duodenal NETs because it is exclusively expressed in these tumors, regardless of their hormonal content and functional activity (124).
TABLE 17.10 Classification of Neuroendocrine Tumors of the Duodenum and Upper Jejunum | |||||||
|---|---|---|---|---|---|---|---|
|
G-cell Tumors (Gastrinomas)
Seventy-five percent of patients have sporadic tumors; 25% associate with MEN-1 (125). These are the most common malignant functional NET. Tumors associated with overt ZES differ from their nonfunctioning counterparts by arising earlier in life in a nonbulbar location and having a higher incidence of metastases.
Ninety percent of gastrinomas arise within the so-called “gastrinoma triangle.” Its superior margin crosses the cystic and common bile ducts, its inferior margin is formed by the junction of the second and third portion of the duodenum, and the medial margin is delineated by the junction of the neck and the body of the pancreas. Approximately 15% of gastrinomas arise in extrapancreatic sites; 13% originate in the second portion of the duodenum. The remainder arise in the stomach, upper jejunum, biliary tree, and lymph nodes within the gastrinoma triangle (126). It has been suggested that precursor cells of gastrin-producing tumors become dispersed during the dorsal rotation of the ventral pancreas during embryonic development and are incorporated into lymphatic tissues (127), thereby providing the anatomic basis for primary nodal gastrinomas. There are also reports of isolated gastrin- or synaptophysin-staining cells in 15% of normal lymph nodes from the gastrinoma triangle (128,129).
Duodenal gastrinomas arising in the setting of MEN-1 are multiple, in contrast to sporadic gastrinomas, which are typically single lesions (130). The multiple tumors seen in MEN-1 patients are thought to be multiple primary lesions based on clonality studies (32). Sixteen percent of duodenal gastrinomas are incidental findings in surgical resection specimens, usually removed for peptic ulcer disease. However, some tumors are large enough to cause clinical symptoms from mass effects, including bile duct obstruction. Approximately one third of patients with gastrinomas have diarrhea. Some patients present with the ZES.
The main features of duodenal gastrinomas include their small size and submucosal location. Most develop in the first and second portions of the duodenum. The tumors may ap-pear slightly polypoid and are often endoscopically misinterpreted as heterotopic pancreas or gastric mucosa or as an inflammatory process. The tumors range in size from 0.2 to 20 cm in diameter (131). However, they measure ≤1 cm in diameter in 62% to 80% of patients. Very small tumors are easily missed during exploratory laparotomy or during gross examination of surgical specimens. Despite their small size, these tumors commonly metastasize and nearly half demonstrate nodal metastases (126,132). Such metastases are often larger than the primary tumors, probably leading to an overdiagnosis of primary nodal gastrinomas.
Gastrinomas display a trabecular/pseudoglandular and/or solid growth pattern. The tumors appear uniform with scanty cytoplasm arranged in solid clusters and ribbons (Figs. 17.25 and 17.26). Some contain a perinuclear collection of whorled microfilaments. Angioinvasion may be present. Gastrinomas
cannot be differentiated from other functional and nonfunctional NETs based solely on their histologic appearance. Gastrin immunoreactivity in the majority of cells establishes the diagnosis. In addition, gastrinomas may produce other polypeptides including glucagon, somatostatin, serotonin, insulin, pancreatic polypeptide, ACTH, enkephalins (133), and hCG.
cannot be differentiated from other functional and nonfunctional NETs based solely on their histologic appearance. Gastrin immunoreactivity in the majority of cells establishes the diagnosis. In addition, gastrinomas may produce other polypeptides including glucagon, somatostatin, serotonin, insulin, pancreatic polypeptide, ACTH, enkephalins (133), and hCG.
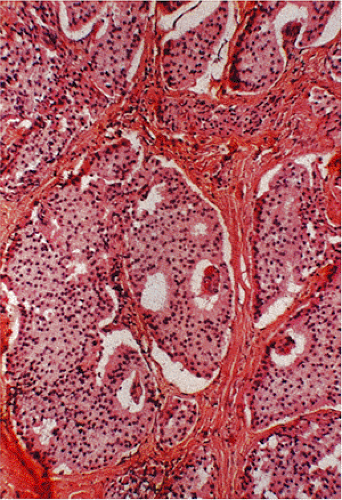 FIG. 17.25. Duodenal gastrinoma found incidentally at the time of resection for a duodenal ulcer. The tumor measured <0.4 mm in diameter. |
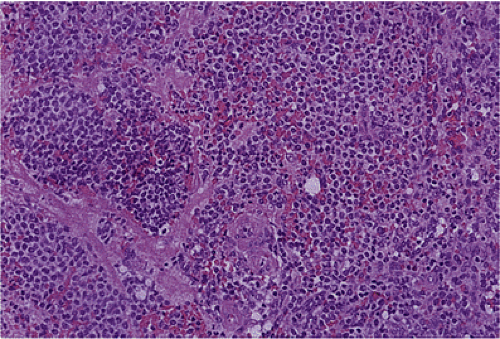 FIG. 17.26. Duodenal gastrinoma resected from the duodenum. Unlike the lesion illustrated in Figure 17.25, this tumor consisted of solid nests of cells with a very monomorphic appearance and not a prominent insular pattern.
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|




