Gastric Cancer: Pathology
Grant N. Stemmermann
Cecilia M. Fenoglio-Preiser
Gastric cancers represent a heterogeneous group of tumors, most lesions being adenocarcinomas. The less common gastric cancers, such as lymphomas, neuroendocrine tumors, and stromal tumors, are discussed elsewhere in this volume.
Gastric adenocarcinoma shows striking temporal, geographic, and subsite variations in frequency, with more than 10-fold differences in incidence existing between countries with high and low rates of the disease (1). High rates are observed in Japan, Korea, and northeast China; in the Andean areas of Latin America; and in Eastern Europe. Once the most common cancer in the United States, gastric cancer distal to the cardia has shown a dramatic decrease in frequency since the 1940s, while the frequency of cancer of the cardia among white American men has more than doubled since 1975 (2). Studies of migrants from high-risk countries, such as Japan, to lower-risk countries, such as the United States, indicate that first-generation migrants continue to experience the high noncardia gastric cancer rates of Japan, whereas their children and grandchildren show rates approaching that of the host country (3). These data support the concept that environmental factors in early life generate precursors to cancer distal to the cardia. These persist into adulthood and are not reversed by living in a more favorable environment.
Gastric adenocarcinoma behaves not as one but as several distinct diseases, depending on the location of the tumor and its histologic type. Gastric cancers may be classified by site of origin into cardiac (gastroesophageal), body, or antral types. Tumors arising in each location differ in their epidemiology, their histology, and their molecular biology. Differences in gastric cancer growth patterns were first recognized by Lauren, who divided gastric cancers into “intestinal” and “diffuse” types based on their histologic characteristics (4). Intestinal cancers have a glandular appearance and expand through the gastric wall, whereas diffuse cancers infiltrate as single discohesive cells and demonstrate an infiltrating growth pattern. The intestinal-type of tumor predominates in populations at high risk of cancer in the distal stomach. This tumor most commonly arises in the antrum in the setting of a multifocal gastritis that is initiated by Helicobacter pylori infection and characterized by extensive atrophy and intestinal metaplasia (5,6,7). Cancers arising in the cardia are also usually of the intestinal type but are not associated with the extensive corpus atrophy that precedes more distal cancers. Diffuse gastric cancers are less common and frequently arise in a nonatrophic corpus that shows H. pylori–induced superficial gastritis. Intestinal-type cancers are more frequent in males and the elderly, and are associated with better survival. Diffuse cancers, in contrast, occur more frequently in women and individuals younger than age 50 (8). They generally have a poor prognosis, and have been associated with blood type A (8) and mutations that inactivate the E-cadherin gene (9,10).
Gastric Cancer Precursors
Gastritis resulting from exposure to environmental hazards in early life precedes the development of cancer distal to the cardia. These include H. pylori infection, a high intake of salt and nitrates, and a diet deficient in fresh fruits and vegetables (11). Middle age, obesity, gastrointestinal reflux disease, and smoking precede the development of cardia cancer (12,13,14).
Gastric carcinomas do not arise de novo from normal gastric epithelium. Like other cancers, gastric carcinomas arise from epithelium that has been altered in some way. The precursor lesions associated with the intestinal-type of antral gastric cancer are well characterized, and include chronic gastritis and intestinal metaplasia. The metaplastic glands resemble those of the small gut, but there are subsite variations in the glycocalyceal enzymes and mucins produced by them. Some metaplastic glands reproduce the cell types of the small bowel. This histologic pattern is termed “complete” metaplasia. Other glands may lack Paneth cells, lose the ability to make some glycocalyceal enzymes, and produce mucosubstances similar to those in the colon, the so-called “incomplete” metaplasia (15). Hybrid goblet cells, sharing both gastric and intestinal phenotypes have also been identified in the metaplastic mucosa (16), as well as gastric and intestinal types of neuroendocrine cells (17). It has been proposed that, in the presence of inflammation, circulating bone marrow–derived stem cells with the potential of differentiating into gastric or intestinal type may take part in gastric cell replication (18). This could account for the observed heterogeneity of intestinal metaplasia. The immunohistochemical labeling indices for cell replication progressively increase with increasing severity of gastritis and are highest in and near areas of intestinal metaplasia. Intestinal metaplasia first appears as discrete foci at the antrocorpus junction (Fig. 21.1). As the process expands, metaplastic glands replace the gastric glands throughout the antrum and extend proximally to replace the acid-producing oxyntic glands of the corpus. The risk of gastric cancer peaks when acid production is at its nadir (Fig. 21.2). Incomplete metaplasia appears in the later stages of this process and is a feature of the extensive metaplasia that results in achlorhydria (7). Serum pepsinogen assays give indirect evidence of the extent of metaplastic replacement of gastric mucosa. Pepsinogen group I (PGI) production is limited to the oxyntic mucosa of the corpus of the stomach, although pepsinogen group II (PGII) is made in all gastric glands. The replacement of oxyntic mucosa by intestinalized mucosa results in decreasing levels of circulating PGI (19). A PGI level <30 ng/mL or a PGI:PGII ratio <2 serve as useful cut points for the identification of extensive intestinal metaplasia, and have been used to identify persons who should be screened because they are at high risk of stomach cancer distal to the cardia (20).
This is especially useful when the PGI level is combined with measurements of the serum H. pylori antibody levels, as shown in Table 21.1, adapted from a recent case-control study (21). Similar trends are observed with PGI:PGII ratios.
This is especially useful when the PGI level is combined with measurements of the serum H. pylori antibody levels, as shown in Table 21.1, adapted from a recent case-control study (21). Similar trends are observed with PGI:PGII ratios.
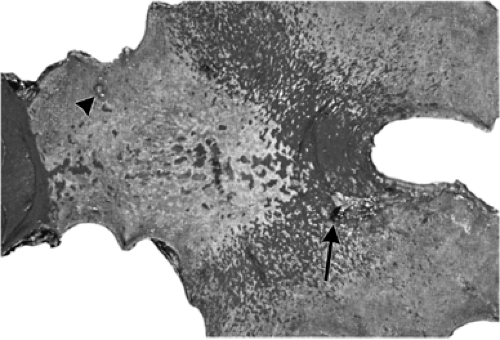 FIGURE 21.1. A gastric resection specimen containing early phase, multifocal gastritis. Foci of intestinal metaplasia are demonstrated by staining for alkaline phosphatase and are limited to the antrocorpus junction and antral lesser curvature. An ulcer (arrow) is present on the posterior wall of the junction zone, and a hyperplastic polyp (arrowhead) is present on the anterior wall of the antrum. |
The mechanisms that underlie in the development of intestinal metaplasia of the gastric mucosa are complex. The maintenance of specific organ structures and functions in the gastrointestinal tract has been attributed to CDX, a homologue of the Drosophila caudal homeobox gene during intestinal development (22). CDX1 and CDX2 are not expressed in the normal stomach, but their mRNAs are expressed in metaplastic glands. CDX2 expression precedes CDX1 expression and appears to be the trigger that initiates the metaplastic process. Mutoh et al. (23) generated transgenic mice that express CDX2 in the gastric parietal cells. By day 37, all gastric mucosal cells are replaced by a full array of intestinal cells, including goblet cells and absorption cells that express alkaline phosphatase. A mechanism for the insertion of this gene into the gastric mucosa is suggested by the recent studies of Houghton and Wang (18). They found that, in the presence of Helicobacter-generated gastritis, circulating bone marrow–derived stem cells may be recruited and engrafted into the replicating zone of the gastric mucosa. This mechanism could account for the observation of hybrid metaplastic epithelium with both gastric and intestinal features in association with H. pylori gastritis (24).
H. pylori infection is closely associated with increased cancer risk, but it does not act alone and not all strains of the organism are equally pathogenic. Strains that possess the cagA gene (25) and those that produce a vacuolating toxin (26) induce a more intense inflammatory reaction, incur greater cell damage, and are associated with greater risk of stomach cancer than other strains. H. pylori infection increases the risk of cancer induction in several ways:
Table 21.1 Age, gender, and ethnicity adjusted odds ratios for combinations of serum PGI levels and Helicobacter pylori antibody status, according to histologic types of gastric cancer | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
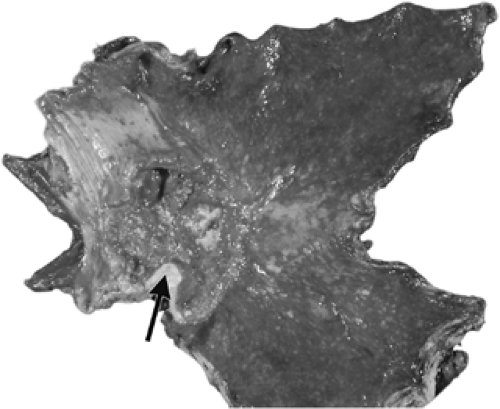 FIGURE 21.2. A resected stomach in which all but a small portion of the greater curvature of the antrum has been converted to alkaline phosphatase–positive intestinalized mucosa. The achlorhydria associated with this change incurs a high risk of carcinoma that, as in this case, characteristically develops at or near the proximal antrum (arrow). |
Its metabolic products may function as direct carcinogens.
More rapid replication of gastric stem cells in response to mucosal injury increases the number of cells vulnerable to ingested carcinogens (Fig. 21.3A, B).
Endogenous byproducts of inflammation, such as superoxide and hydroxyl radicals, may initiate mutations through oxidative damage.
The appearance of a bacterial flora with the capacity of generating carcinogenic nitroso compounds from ingested amines is a feature of the achlorhydria, whether associated with end-stage H. pylori infection or pernicious anemia (27).
Fifty percent of patients with gastric juice pH >4 have nitrate-reducing bacteria in their stomachs (28).
H. pylori attach to the surface epithelium (Fig. 21.4) but produce cell damage in the area of the mucus neck region, which contains actively replicating cells (Fig. 21.5). The inflammatory cells generate free radicals and reactive nitrogen
molecules that are capable of damaging cellular DNA and presumably increase gastric cancer risk through induction of mutations in genes critical to cell division and repair.
molecules that are capable of damaging cellular DNA and presumably increase gastric cancer risk through induction of mutations in genes critical to cell division and repair.
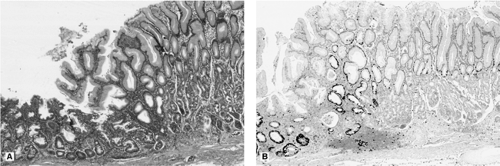 FIGURE 21.3. A: A hematoxylin-and-eosin–stained section has a focus of intestinal metaplasia (left). The adjacent gastric mucosa is hyperplastic. B: Ki-67 immunohistochemistry reflects the increased cell replication in the metaplastic mucosa and the gastric mucosa adjacent to it. |
H. pylori gastritis evolves from an acute to a chronic gastritis that is characterized by a polymorphic infiltrate in the upper lamina propria, consisting of neutrophils, eosinophils, basophils, monocytes, and plasma cells (Fig. 21.6). Foci of atrophy associated with lymphocytic follicles and isolated metaplastic glands follow (Fig. 21.7).
Mucosal inflammation also precedes other types of gastric cancer. As noted previously, diffuse carcinomas arise in patients whose stomachs show intense, H. pylori–induced superficial gastritis. The risk of acquiring diffuse cancer is especially high when CagA is the infecting strain (21). The onset of diffuse cancers in the nonatrophic corpus of young patients suggests an increased vulnerability to cancer induction, either from an intense immunologic response to infection, from infection by toxic strains of H. pylori, or from greater exposure to exogenous carcinogens. Inherited mutations or polymorphisms may also contribute to this increase in risk.
Gastric Ulcer
In the early stages of the expansion of H. pylori–induced multifocal gastritis, the intestinalized mucosa at the antrocorpus junction is exposed to acid and pepsin produced by the still intact oxyntic mucosa. The intestinalized mucosa lacks the protective mucus barrier of the normal stomach and is therefore vulnerable to peptic ulceration. Patients with chronic gastric ulcers are at greatly increased risk of gastric cancer. Re-epithelialization of the ulcerated surface associates with expansion of the replication zones in the intact glands adjacent to the ulcer (29). This exposes a larger number of replicating cells to genotoxic damage and accounts for the observation that dysplasia and early cancers arise in the gastric mucosa bordering ulcers, rather than in the ulcer itself (30). An example of this phenomenon is shown in Fig. 21.8A and B.
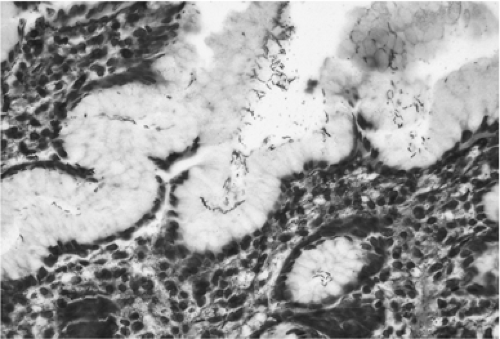 FIGURE 21.4. A Steiner stain shows the characteristic superficial location of Helicobacter pylori organisms in the infected stomach. |
Autoimmune Gastritis
Autoimmune gastritis, like environmental gastritis, increases the risk of gastric cancer. Patients with autoimmune gastritis produce autoantibodies to parietal cell membranes (31,32,33), intrinsic factor, and the gastrin receptor (34). Patients show progressive destruction of the acid-secreting mucosa, ultimately leading to hypochlorhydria or achlorhydria. Inflammation centering on the fundic glands is characterized by the accumulation of lymphocytes and plasma cells in the lamina propria. Depending on the stage of the disease, variable degrees of parietal and chief cell loss are present. The sustained inflammatory process
and the gastrin-induced growth promotion that accompanies achlorhydria results in increased cell replication. The foveolar epithelium is unaffected by this process and may even be hyperplastic (Fig. 21.9), sometimes to the point of forming hyperplastic polyps (Fig. 21.10A, B). Eventually, the fundic mucosa is completely replaced by pyloric and, later, by intestinalized glands. As in end-stage environmental gastritis, the achlorhydric stomach favors the generation of a bacterial flora that may generate carcinogenic nitroso compounds. The areas of intestinal metaplasia may undergo dysplastic alterations that can progress to invasive cancer.
and the gastrin-induced growth promotion that accompanies achlorhydria results in increased cell replication. The foveolar epithelium is unaffected by this process and may even be hyperplastic (Fig. 21.9), sometimes to the point of forming hyperplastic polyps (Fig. 21.10A, B). Eventually, the fundic mucosa is completely replaced by pyloric and, later, by intestinalized glands. As in end-stage environmental gastritis, the achlorhydric stomach favors the generation of a bacterial flora that may generate carcinogenic nitroso compounds. The areas of intestinal metaplasia may undergo dysplastic alterations that can progress to invasive cancer.
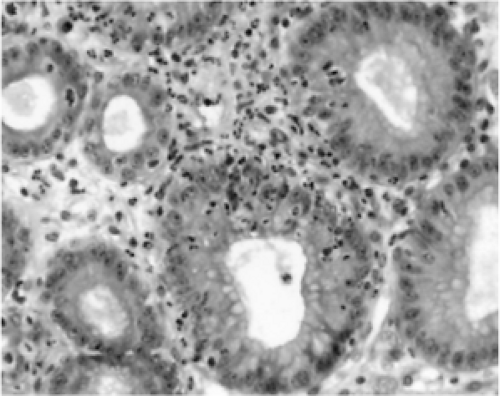 FIGURE 21.5. A hematoxylin-and-eosin stain showing a characteristic tissue reaction to Helicobacter pylori infection—mononuclear infiltration of the replication zones of the gastric glands. |
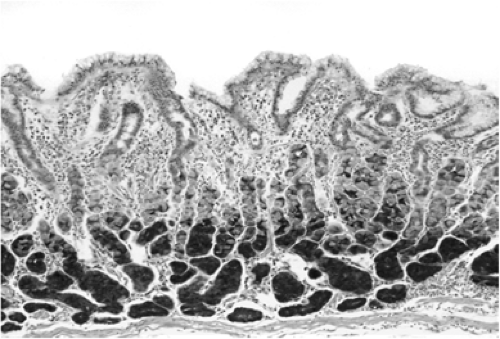 FIGURE 21.6. An immunohistochemical study of an antrum containing the superficial gastritis that characterizes the early phases of Helicobacter pylori gastritis. The pepsinogen group II–expressing antral glands are not yet atrophic. |
Reflux Gastritis
Long-term exposure to refluxed bile salts and pancreatic juice in the gastric remnant after subtotal gastrectomy elicits mucosal inflammation. This associates with increased risk of cancer in the gastric remnant starting at 17 years after the resection (35). Reflux of bile and pancreatic juice may also occur in patients with an intact stomach, and this has been associated with an increased risk of gastric and gastroesophageal cancer (36).
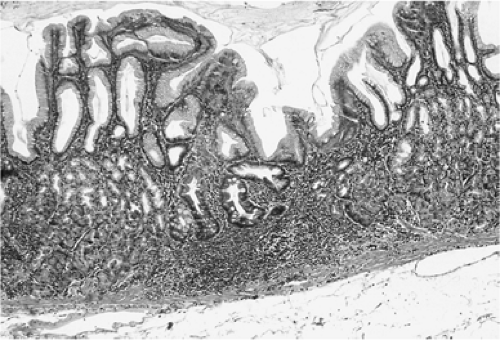 FIGURE 21.7. A hematoxylin-and-eosin–stained section of stomach contains an early focus of atrophy and intestinal metaplasia in the oxyntic mucosa of the corpus. These foci typically consist of one or two intestinalized glands and are associated with a lymphocytic follicle. |
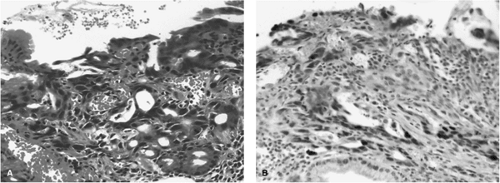 FIGURE 21.8. A: Hematoxylin-and-eosin–stained ulcer margin has a small area of dysplastic mucosa bordering a healing ulcer. B: This dysplastic mucosa has p53 overexpression shown by immunohistochemistry (See also color Figure 21.8a,b). |
Dysplasia
Dysplasia represents intraepithelial neoplasia and is the immediate precursor to invasive cancer. The dysplastic cells may be either gastric or intestinal in type, or may be hybrid forms from each lineage. Dysplastic gastric cells may be encountered at the margins of chronic gastric ulcers, while the intestinal forms predominate in stomachs showing confluent intestinal metaplasia. Both forms associate with immunohistochemical overexpression of p53 (Figs. 21.8B and 21.11). The dysplastic mucosa may be polypoid or flat, whether intestinal or gastric in type. The dysplastic cells show increased nuclear size, hyperchromasia, and irregularity in shape. These cytologic changes may be superimposed on a disorganized mucosal architecture.
The World Health Organization (WHO) classification for gastric dysplasia divides it into mild, moderate, and severe
categories (37,38), but we believe that it is sufficient to classify dysplasia as either high or low grade. Dysplasia may be limited to the upper third of the mucosa, as shown in Fig. 21.11, or may involve the full thickness of the mucosa (Fig. 21.12). Correa described the now widely accepted sequence of steps that begins with gastritis, leading to intestinal metaplasia, then dysplasia, and ultimately to invasive gastric cancer (39), as shown in Fig. 21.13. Both genetic (40,41) and epigenetic (42,43) alterations increase in frequency with each step from the normal mucosa to gastritis, to metaplasia, to dysplasia, and to invasive cancer.
categories (37,38), but we believe that it is sufficient to classify dysplasia as either high or low grade. Dysplasia may be limited to the upper third of the mucosa, as shown in Fig. 21.11, or may involve the full thickness of the mucosa (Fig. 21.12). Correa described the now widely accepted sequence of steps that begins with gastritis, leading to intestinal metaplasia, then dysplasia, and ultimately to invasive gastric cancer (39), as shown in Fig. 21.13. Both genetic (40,41) and epigenetic (42,43) alterations increase in frequency with each step from the normal mucosa to gastritis, to metaplasia, to dysplasia, and to invasive cancer.
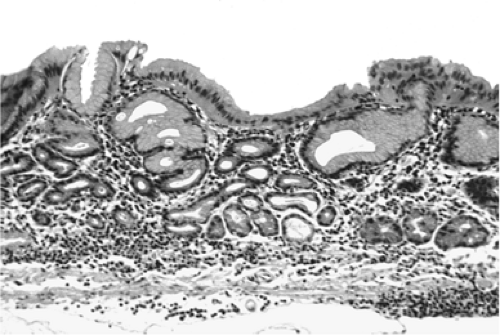 FIGURE 21.9. Severe mucosal atrophy of the corpus in a patient with autoimmune gastritis. Neither parietal cells nor corpus chief cells remain. The stomach lining consists of a somewhat hyperplastic foveolar epithelium and actively replicating neck zones. A uniform lymphocytic infiltrate occupies the lamina propria. |
Progression of Dysplasia to Invasive Cancer
The risk of progression of dysplasia to cancer is highest in individuals with moderate to severe dysplasia (44). One study of 93 cases of gastric dysplasia showed progression to a more severe grade of dysplasia or evolution to invasive cancer in 21% of low-grade dysplasias, 33% of moderate dysplasias, and 57% of severe dysplasias (45). Dysplasia progresses slowly, usually over a period of months to years (44). However, it may also remain stable or even regress (46,47). A high percentage of closely followed cases of severe dysplasia ultimately develop into invasive carcinoma (47).
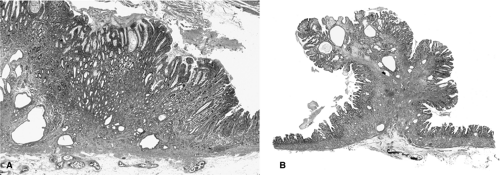 FIGURE 21.10. A: A hyperplastic polyp in the corpus in a patient with autoimmune gastritis. B: An antral hyperplastic polyp in this patient. |
Gastric Adenomas
Some forms of gastric dysplasia present as sessile or pedunculated adenomas closely resembling colorectal tubular adenomas (Fig. 21.14). Gastric adenomas and carcinomas have similar epidemiologic backgrounds, are more commonly identified in the antrum or the midstomach than in the corpus, and arise in association with intestinal metaplasia (48). Only a minority of early gastric cancers arises in adenomas, but adenomas are clearly precancerous and their treatment must be aimed at total removal (49).
Hyperplastic Polyps
Hyperplastic polyps represent the most commonly encountered polyps in the stomach (49). These lesions often develop adjacent to gastric remnants, ulcers, or gastroenterostomy stomas. They also commonly arise in stomachs with chronic gastritis. Hyperplastic polyps are exuberant regenerative responses of the gastric foveolar cells rather than true neoplasms, but hyperplasia may coexist with either adenoma or carcinoma in the same polyp (49). The synchronous appearance of hyperplastic polyps and gastric cancers may merely reflect the propensity of both lesions to arise in a setting of chronic gastritis or atrophy.
Fundic Gland Polyps
Fundic gland polyps were initially described in patients with familial adenomatous polyposis (FAP) (48). Fundic gland polyps also develop in the absence of familial polyposis and usually affect individuals in middle age. Most fundic gland polyps do not have malignant potential, but those found in FAP may be dysplastic.
Histogenesis of Gastric Cancers
Little is known about the exact cell of origin of gastric carcinomas. Many tumors arise on a background of chronic atrophic gastritis with intestinal metaplasia. Metaplastic changes in the mucosa adjacent to intestinal-type carcinomas are quite common but are less frequent in diffuse-type cancers (50,51). As discussed previously, analysis of early intestinal-type carcinomas demonstrates that areas of intestinal metaplasia commonly
occur adjacent to the tumors. In contrast, in small diffuse-type cancers, the neoplastic cells form an intimate association with the replication zone in the neck of the gland, suggesting that they originate from the undifferentiated cells residing in this region (52).
occur adjacent to the tumors. In contrast, in small diffuse-type cancers, the neoplastic cells form an intimate association with the replication zone in the neck of the gland, suggesting that they originate from the undifferentiated cells residing in this region (52).
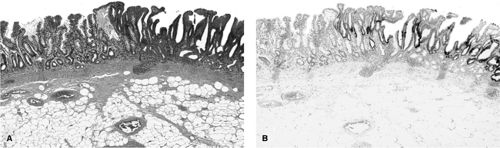 FIGURE 21.11. A: A hematoxylin-and-eosin–stained section of stomach containing confluent intestinalization. A well-defined area of flat dysplasia is characterized by cells with large dark nuclei and loss of mucus production. B: p53 Overexpression, shown by immunohistochemistry, highlights the dysplastic mucosa. |
Identification of a single cell of origin is often difficult in gastric cancers. Gastric carcinomas are heterogeneous lesions that may contain numerous cell types, including those with features of intestinal columnar cells (53,54,55), intestinal goblet cells, pyloric mucous cells (54,55,56), foveolar cells (53,54), Paneth cells (56), parietal cells (57,58), squamous cells (54), and endocrine cells (59,60). In addition, numerous hybrid cell forms occur (53). The identification of multiple cell types in gastric cancers probably reflects the fact that most of the gastric epithelial cells originate from a single multipotential progenitor cell.
Topography of Gastric Cancer
Gastric carcinomas in high-risk populations most commonly arise on the lesser curvature of the antrum and are especially common at mucosal junctional areas, such as the prepyloric region and incisura on the lesser curvature at the antrocorpus junction (7). Less common sites of origin include the cardia (2,61) and the greater curvature of the corpus (7). Identification of the point of origin of an esophageal adenocarcinoma or cancer of the cardia is not always possible (62). Some observers favor the designation of “gastroesophageal junction tumors” to these cancers because the two tumors share similar epidemiologic backgrounds, recent increases in frequency among middle-age white males, an association with intestinal metaplasia of the distal esophagus, and similar molecular features (62,63). Neither cancer associates with H. pylori gastritis nor the multifocal gastritis that the infection produces. It has even been suggested that H. pylori could be protective against these tumors (64,65).
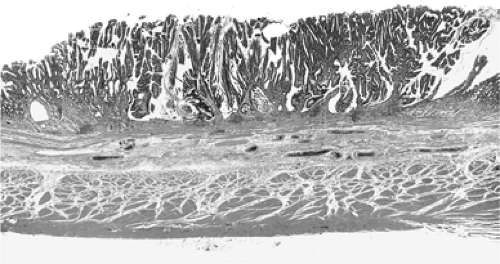 FIGURE 21.12. A hematoxylin-and-eosin–stained section of full-thickness dysplastic intestinalized mucosa. The lesion resembles a colonic villous adenoma. |
Early Gastric Cancer
Gastric carcinomas are divided into early and advanced stages, depending on the depth of invasion into the gastric wall. Early gastric carcinoma is defined as a cancer that remains limited to the mucosa or the submucosa, regardless of the presence or absence of nodal metastases. The subtypes of early cancer, first defined in Japan, are now generally accepted worldwide (Fig. 21.15) (66).
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


