Gastric Cancer: Molecular Biology and Genetics
Rajnish Mishra
Steven M. Powell
Introduction
General Epidemiology
Gastric carcinoma remains one of the world’s most common cancers and a leading cause of cancer death worldwide (1). Global estimates suggest that 933,937 new cases of gastric carcinoma will be diagnosed and 700,349 will die from this disease each year (2). The American Cancer Society projects that 21,760 new cases of gastric cancer will be diagnosed in the United States this year (2). Neoplasia of the stomach is largely composed of adenocarcinomas, accounting for more than 95% of cases. Primary lymphoma, stromal tumors (i.e., leiomyosarcoma, liposarcoma), and carcinoid tumors can also arise in the stomach, but malignant tumors of these types do not occur as often.
Interestingly, a sharp increase in incidence of gastroesophageal junction adenocarcinomas was recently noted in the white male population of the United States as well as in other populations (3,4). Significant geographic variability in incidence both intra- and internationally is observed (5). Epidemi-ologic studies that include migration and temporal analyses indicate that environmental factors, especially in the first decades of life, are important in the etiology of gastric cancers (6,7). Notably, a consistent predominance (2:1 ratio) of gastric cancers in males is seen worldwide. With such a temporal and regional variation in gastric cancer incidence rates, a better understanding of these phenomena through molecular studies of gastric tumorigenesis will provide important insights into cancer development in general, and it is anticipated to lead to earlier diagnosis and better management options in combating this devastating cancer. In addition, multiple gastric tumor pathological classification systems have been formulated in efforts to identify various subgroups with different biological behavior and prognostic indicators. Molecular markers should help facilitate the classification of the various subgroups of gastric carcinomas.
Helicobacter pylori infection appears to lead to a five- to sixfold increase in risk of gastric malignancy, both adenocarcinoma and primary non-Hodgkin lymphoma (8,9). Yet, most of those infected with this micro-organism do not develop gastric cancer. Evidence of Epstein-Barr virus infection has been noted in a small proportion of gastric carcinomas, especially those that exhibit lymphoepithelial histopathology. The relative importance of bacterial virulence factors, environmental factors, and host factors (i.e., age of acquisition, immune responses, acid secretion changes) involved in the clinical outcome of these infections and other environmental exposures are currently pressing issues, and molecular studies may help discern the true influential factors. Indeed, Park et al. noted that gene–environment interactions between CYP2E1 gene polymorphism and smoking may alter the susceptibility for cancer development in the stomach (10).
Host Observations
A population association study recently demonstrated the more frequent occurrence (54%) of the human leukocyte antigen (HLA) DQB1*0301 allele in Caucasian patients with gastric cancer than in a control noncancerous group (27%) (11). If confirmed not to represent ethnic heterogeneity, this association may imply that this locus itself directly influences susceptibility to gastric cancer development or is a marker in linkage disequilibrium with a nearby cancer-predisposing locus. In another study, the frequency of allele DQB1*0401 was significantly higher in those infected with H. pylori who developed atrophic gastritis than in those who were infected and did not develop atrophic gastritis or those not infected (12). The potential role of the HLA locus in gastric tumorigenesis has implications for the importance of a potential escape mechanism from immune surveillance as a causative factor for this disease. The fact that most people with these alleles do not develop gastric cancer illustrates the complexity of this multifactorial disease.
Of note, the blood group A phenotype was reported to be associated with gastric cancers in the 1950s (13,14). Interestingly, H. pylori was shown to adhere to the Lewis blood group antigen, indicating a potentially important host factor that might facilitate this chronic infection and subsequent risk of gastric cancer (15). In addition, small variant alleles of a mucin gene, Muc1, were found to be associated with gastric cancer patients compared to a blood donor control population (16). Confirmation or studies demonstrating the relevance of these findings are still awaited. In fact, a recent study found no correlation between blood group phenotype including A and Lewis and the occurrence of gastric cancer (17).
Several precursor lesions have been identified in the development of intestinal-type gastric carcinomas (18). Dysplasia is generally regarded as a true precancerous lesion; however, the magnitude of atrophic gastritis and intestinal metaplastic (incomplete type) lesions as precancerous lesion remains to be defined. Although an association of chronic atrophic gastritis and gastric intestinal metaplasia has been observed in patients with gastric carcinoma, most individuals harboring these lesions do not develop gastric cancers (19). Furthermore, a low serum pepsinogen 1-to-pepsinogen 2 ratio has been used to identify patients harboring gastric atrophy (20).
Familial Epidemiology
Most cases of gastric cancer appear to occur sporadically without an obvious inherited component. It is estimated that as many as 8% to 10% of gastric cancer cases are related to an inherited familial component (21). Familial clustering has been observed in 12% to 25% of gastric carcinoma cases and dominant inheritance patterns observed (22,23). Notably, Napoleon Bonaparte was apparently afflicted with gastric cancer involving most of his stomach and may have also had other family members (e.g., father, sister) afflicted with this cancer (24,25). In the Swedish Family Cancer Database, which is the largest study of familial gastric cancer published to date, the standardized incident rates were 1.31 (95% confidence interval [CI], 0.97–1.70) and 1.7 (95% CI, 1.08–1.92) when a patient presented with gastric carcinoma. Risk was 1.59 (95% CI, 1.10–2.16) in offspring whose diagnosis was at an age younger than 50 years (26).
Case-control studies have observed consistent, up to threefold increases in risk for gastric cancer among relatives of gastric cancer patients (23,27). A population-based control study found increased risk for gastric cancer when first-degree relatives were affected (OR = 1.7 with a parent, OR = 2.6 with a sibling), with the risk increasing (OR up to 8.5) if more than one first-degree relative was affected (28). Interestingly, a higher risk was noted in individuals with an affected mother versus an affected father. Studies of monozygotic twins have even shown a slight trend toward increased concordance of gastric cancers compared to dizygotic twins (29,30).
Several genetic susceptibility traits with an inherited predisposition to gastric cancer development are described in this chapter. A few are well-characterized inherited predisposition syndromes potentially involving gastric cancer development, such as hereditary nonpolyposis colorectal cancer (HNPCC). Other clinical entities with a predisposition to gastric tumorigenesis are just being unveiled, such as kindreds manifesting diffuse, poorly differentiated gastric cancers and cosegregating germline E-cadherin alterations.
Inherited Genetic Susceptability
E-cadherin in Diffuse Gastric Cancers
Large families with an obvious autosomal dominant, highly penetrant inherited predisposition for the development of gastric cancer, having the potential power to link disease markers, are rare. A large Maroi kindred manifesting early onset diffuse gastric cancers was investigated for linkage analysis, and disease was found to be linked to the E-cadherin/CDH1 locus on 16q and associated with significant mutations in this gene (31). Since then, more than 14 truncating germline E-cadherin mutations have been reported scattered across 8 of the 16 exons this gene encompasses (31,32,33,34,35,36). The age of onset and diagnosis of diffuse gastric cancer in those who harbored germline mutations of E-cadherin ranged from 14 to 69 years of age. The incomplete penetrance of germline E-cadherin mutation was seen with obligatory carriers who remained unaffected in their eighth and ninth decades of life. One of the larger studies of ten kindreds manifesting diffuse gastric cancers identified three families with germline E-cadherin mutations (32). No germline mutations of this gene were detected in apparent “sporadic” diffuse gastric cancer cases in Britain with a mean age of 62 years (35).
Based on the results of E-cadherin mutation, screening of the probands from 42 new families with diffuse gastric cancers, Brooke-Wilson et al. determined that 40% of families with multiple gastric cancers and at least one diffuse gastric cancer diagnosed in an individual younger than 50 years had a pathological germline E-cadherin mutation and thus recommended it as a screening criteria (37). Therefore, genetic counseling for those kindreds manifesting a strong predisposition for the development of diffuse gastric cancer is imperative in their medical management (Fig. 20.1). The diffuse familial gastric cancer families are one of the subgroup of kindreds for which genetic testing can now be offered. It is noteworthy though that two-thirds of hereditary diffuse gastric cancer families reported to date have proved negative for E-cadherin gene mutation (37).
Hereditary Nonpolyposis Colorectal Cancer
A now well-characterized inherited predisposition syndrome that can involve gastric cancer development is HNPCC (38). Germline genetic abnormalities of mismatch repair (MMR) genes underlying this disease entity have been unveiled and include potential tumor development in a variety of tissue types (39). Gastric carcinomas that occur in this setting were predominantly diagnosed at the mean age of 56 years, of the intestinal type, without H. pylori infection detected, and most exhibited microsatellite instability (MSI) in a Finnish HNPCC registry study (40). Renkonen-Sinisalo et al. studied gastric histopathology comparing 73 mutation-positive and 32 mutation-negative families for difference in occurrence of H. pylori, atrophy, inflammation, intestinal metaplasia (IM), and dysplasia. They identified only a single case of duodenal cancer among the mutation-positive individuals, but there was no evidence of gastric neoplastic lesions in either group. Thus, abnormalities of MMR genes rarely cause gastric cancer (41).
Interestingly, the occurrence of gastric cancers associated with HNPCC has decreased, similar to the recent general decline in incidence of gastric cancer in developed countries (42). The isolation and characterization of these predisposing gene alterations should allow better definition of the fraction of gastric cancers that result from this trait. Testing for MSH2 and MLH1 gene alterations is currently readily available and can be used to identify those gastric cancer cases that are part of this specific cancer predisposing trait.
Other Inherited Traits
There has even been a report of gastric carcinoma in an extended Li-Fraumeni kindred with an underlying p53 germline alteration (43). Gastric cancers have also been noted to occur in patients with gastrointestinal polyposis disease entities such as familial adenomatous polyposis (FAP) and Peutz-Jeghers syndrome (PJS) (44,45). Interestingly, an increased risk of gastric cancer associated with FAP (patients with germline APC mutations) has been reported in high-risk regions such as Asia (46), whereas no increased risk was exhibited in other populations (47). Overall, gastric carcinoma is rare in these settings, and the exact contribution of the polyposis and underlying germline alterations of APC (FAP) and LKB1/STK11 (PJS) to gastric cancer development in these cases is unclear.
Rare kindreds exhibiting site-specific gastric cancer predi lection have been reported, occasionally associated with other inherited abnormalities (48,49). A constitutional deletion of 18p inherited from her mother with subsequent loss of 18q and the whole chromosome was observed in the gastric carcinoma of a 14-year-old patient with associated mental and cardiac abnormalities, suggesting a predisposing condition (50). A kindred manifesting the autosomal dominant inheritance of familial gastric polyposis and gastric cancer development was demonstrated not to have a germline E-cadherin mutation,
and on linkage analysis, was not linked to 16q, the locus of E-cadherin (32,51). Thus, other loci appear to exist that when altered may predispose an individual to gastric cancer development.
and on linkage analysis, was not linked to 16q, the locus of E-cadherin (32,51). Thus, other loci appear to exist that when altered may predispose an individual to gastric cancer development.
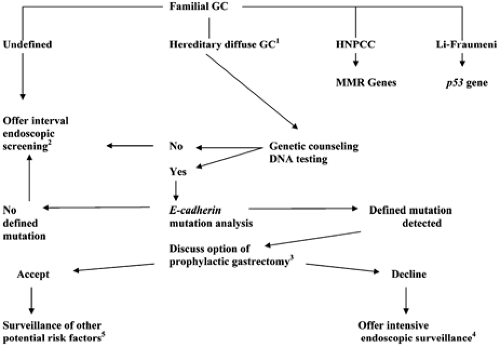 FIGURE 20.1. Algorithm for guidance in managing familial gastric cancer (GC) kindreds. Those identified to meet clinical criteria for hereditary nonpolyposis colon cancer syndrome (HNPCC) and the Li-Fraumeni syndrome are counseled for consideration of genetic testing. MMR, mismatch repair. Microsatellite instability testing can aid in diagnosing HNPCC. 1This algorithm was initially formulated at the consensus symposia where clinical criteria were generated to define hereditary diffuse gastric cancer: two or more cases in first/second-degree relatives, at least one diagnosed before the age of 50 years, or three or more cases of documented diffuse gastric cancer in first/second-degree relatives (33). 2Consider individual age-dependent familial expression to determine the initiation, interval, and intensity of screening exams. Prophylactic gastrectomies have been performed in kindreds expressing highly penetrant phenotypes without prior knowledge of causative mutations. 3Consider age-dependent familial expression in determining management. 4Endoscopic ultrasound and chromographic (methylene blue or indigo carmine staining) endoscopy can be applied in attempts to increase the sensitivity of detecting early lesions in the stomach. 5One should have a high index of suspicion for the potential development of other cancers such as that of the breast, colon, and endometrium. |
Somatic Molecular Genetics
General
Most molecular analyses of this cancer have involved studies of sporadic tumors for critical, acquired alterations. A detailed, clear working model of gastric tumorigenesis has yet to be formulated. Multiple somatic alterations have been described, but the significance of these changes in gastric tumorigenesis remains to be established in most instances. Molecular studies may provide new avenues for lowering the dismal gastric cancer mortality rate.
Cytogenetic studies of gastric adenocarcinomas are few in number, with a variable number of numerical or structural aberrations, and have failed to identify any consistent or noteworthy chromosomal abnormalities (52,53). Comparative genomic hybridization analyses of xenografted and primary gastric and gastroesophageal junctional adenocarcinomas have revealed several regions of consensus change in DNA copy number that may indicate the location of candidate oncogenes and tumor suppressor genes involved in gastric tumorigenesis (54,55). Chromosomal arms 4q, 5q, 9p, 17p, and 18q showed frequent decreases in DNA copy number. However, chromosomes 7, 8, and 20q showed frequent increases in DNA copy number of cases analyzed in this fashion. Loss of heterozygosity (LOH) analyses have identified several arms and regions of chromosomes that contain or potentially harbor tumor suppressor genes important in gastric tumorigenesis, including 17p (>60% at p53’s locus) (56), 18q (>60% at SMAD4 and DCC’s loci) (57), and 5q (30%–40% at or near APC’s locus) (56,58). Comprehensive LOH analysis in xenografted adenocarcinomas identified 3p, 4p, 4q, 5p, 8q, 13p, 17p, and 18q to be frequently lost well above background (59). In LOH analysis of more than 100 archived stomach cancers, allelic loss was most frequently noted on chromosome 3p (60). Moreover, three distinct regions of chromosome 4q were found to be frequently lost in gastroesophageal junctional adenocarcinomas, indicating the potential of multiple tumor suppressor genes on this chromosomal arm (61).
Notable allelic loss of both 17p and 18q in proximal or gastroesophageal junctional tumors was associated with a poorer survival than those cases without allelic loss or just one allelic loss at their loci (62). Knowledge of these critical alterations is important because gastric cancers of different histopathological features have been shown to be associated with distinct patterns of genetic alterations, supporting the notion that they evolve through distinct genetic pathways. As described later in
this section, both known and candidate tumor suppressor genes have been isolated in some of these frequently lost regions, but the actual targets of genetic loss that provide gastric neoplastic cells with additional survival or growth advantages for clonal expansion remain to be clarified for many of these loci.
this section, both known and candidate tumor suppressor genes have been isolated in some of these frequently lost regions, but the actual targets of genetic loss that provide gastric neoplastic cells with additional survival or growth advantages for clonal expansion remain to be clarified for many of these loci.
Instability: Chromosomal and Microsatellite
The majority of gastric cancers exhibit significant gross chromosomal aneuploidy. One study found that 72% of differentiated tumors and 43% of undifferentiated gastric tumors were aneuploid (63). However, MSI has been found in a subset of sporadic gastric carcinomas ranging from 13% to 44% of tumors (64). Variability in classification of instability or histopathological subtype and number of loci examined in studies account for some variation of this phenotype’s frequency, with a trend toward more frequent occurrence in intestinal-type cancers at more advanced stages observed. The degree of genomewide instability also varies with more significant instability (e.g., microsatellite instability-high [MSI-H] exhibiting >33% unstable loci of those tested) occurring in only 10% to16% of gastric cancers (65).
Alterations responsible for producing the MSI-H phenotype in a subset of sporadic gastric cancers have been elucidated. Abnormal loss of protein expression of either MLH1 or MSH2 was demonstrated in all cases exhibiting MSI-H (66). Altered expression of MLH1 was associated with increased methylation of the promoter region of MLH1 in MSI-H cases, suggesting a silencing role of hypermethylation (67,68). Distinct methylation of the promoter of hMLH1 was noted in five of eight MSI-H cases, whereas none of 43 microsatellite instability-low (MSI-L) or microsatellite stable (MSS) cases exhibited this methylation (69).
MSI-H gastric tumors exhibit distinct clinicopathological characteristics. Consistent associations of the MSI-H phenotype with intestinal subtype, distal location (e.g., antral), and more favorable prognosis have been observed (65,70,71,72,73,74). In addition, some but not all studies have noted associations between the MSI-H phenotype and less frequent lymph node metastasis (65,71,72), greater depth of invasion (72), near-diploid DNA content (65), and tumoral lymphoid infiltration (65,71,72). A possible explanation for the unique clinicopathological phenotype observed in MSI-H gastric tumors may be the occurrence of mutations in a distinct set of cancer-related genes differing from those in tumors with no or low-level MSI. Because MSI-H gastric carcinomas appear to be clinicopathologically distinct, it may ultimately prove valuable to have markers that identify this subgroup of gastric cancers, such as BAT-26 (66). Several tumor suppressor genes have been shown to be critical targets of defective MMR in MSI-H tumors. Moreover, a proapoptotic gene and additional MMR genes have been demonstrated to be altered in MSI gastric cancer cases. These same genes are observed to be infrequently mutated or altered in MSI-L or MSS tumors (71,75,76).
At least one important target of MSI appears to be the transforming growth factor (TGF)-β type II receptor (TGFβR2) at a polyadenine tract within its gene (77). Altered TGFβR2 could also be found in gastric cancers not displaying MSI. Several gastric cancer cell lines resistant to the growth inhibitory and apoptotic effects of TGF-β were shown to have abnormal TGFβR2 genes and/or transcripts (78). Moreover, some gastric cancer cell lines and 5 of 40 primary gastric cancers (12.5%) exhibited hypermethylation of the promoter region of TGF-β type 1 receptor gene and decreased mRNA expression (79). Another gene involved in this signaling pathway, ACVR2, was found to be similarly mutated, even in a biallelic fashion, in gastric tumors exhibiting MSI (80). Thus, alteration of TGF-β receptors and other members of this signaling path appears to be a critical event in the development of at least a subset of gastric cancers, allowing escape from the growth control signal of TGF-β.
Additional genes with simple tandem repeat sequences within their coding regions found to be altered in gastric cancers displaying MSI include BAX, IGFRII, and E2F-4, which are known to be involved in regulation of cell cycle progression and apoptotic signaling (81,82,83,84). Furthermore, somatic nonframeshift mutations have been reported in BAX (81). Moreover, the relatively frequent missense mutations at codon 169 of BAX were shown to impair its proapoptotic activity (85).
Specific Alterations
The p53 gene has consistently been demonstrated to be signifi cantly altered in gastric adenocarcinomas. Allelic loss occurs in more than 60% of cases, and mutations are identified in approximately 30% to 50% of cases, depending on the mutational screening method employed and variable sample sizes (86). Some mutations of p53 have even been identified in early dysplastic and apparent intestinal metaplasia gastric lesions; however, most alterations occurred in the advanced stages of neoplasia. The spectrum of mutations in this gene within gastric tumors is not unusual with a predominance of base transitions, especially at CpG dinucleotides. Many studies have used immunohistochemical analysis of tumors in an effort to detect excessive expression of p53 as an indirect means to identify mutations of this gene, but this assay does not appear to have consistent prognostic value in patients with gastric cancers (87,88).
Several sporadic gastric cancers have displayed altered E-cadherin, mainly diffuse types. E-cadherin is a transmembrane, calcium ion-dependent adhesion molecule important in epithelial cell homotypic interactions that, when decreased in expression, is associated with invasive properties (89). Reduced E-cadherin expression determined by immunohistochemical analysis was noted in the majority (92% of 60 cases) of gastric carcinomas when compared to the adjacent normal tissue (90). Furthermore, absent expression of E-cadherin was observed to be significantly associated with undifferentiated, diffuse-type cancers (n = 30) compared to intestinal-type cancers (n = 30). Genetic abnormalities of the E-cadherin gene (located on chromosome 16q22.1) and transcripts were demonstrated in 13 of 26 diffuse gastric cancers on reverse transcriptase polymerase chain reaction (RT-PCR) analysis (91). Moreover, a study of ten gastric cancer cell lines displaying loose intracellular adhesion found absent E-cadherin transcripts in four lines, and insertions or deletions in two other lines (92). E-cadherin splice site alterations producing exon deletion and skipping, large deletions including allelic loss and point mutations, mostly of the missense nature, have been demonstrated in diffuse-type cancers, some even exhibiting alterations in both alleles (93). Seven of ten diffuse-type gastric carcinomas were found to contain somatic E-cadherin alterations, including the diffuse component in five of six mixed-type tumors (94). Methylation of the promoter region of E-cadherin was found in 16 (26%) of 61 gastric cancers studied (69). In addition, α-catenin, which binds to the intracellular domain of E-cadherin and links it to actin-based cytoskeletal elements, was noted to have reduced immunohistochemical expression in 70% of 60 gastric carcinomas and correlated with infiltrated growth and poor differentiation (95).
Evidence of tumor suppressor loci on chromosome 3p has accumulated from a variety of studies, including allelic loss in primary gastric tumors (46%) and homozygous deletion in a gastric cancer cell line (KATO III) as well as xenografted tumors (96). The FHIT gene was recently isolated from the common fragile site FRA3B region at 3p14.2 and found to have abnormal transcripts with deleted exons in five of nine
gastric cancers (97). Furthermore, loss of FHIT protein expression was demonstrated immunohistochemically in the majority of gastric carcinomas (98). One somatic missense mutation was identified in exon 6 of the FHIT gene during a coding region analysis of 40 gastric carcinomas (99). Additional studies are needed to identify the critically altered targets on this chromosome, clarify the role FHIT plays in gastric tumorigenesis, and determine the role breakpoints in this region of 3p have in gastric cancer development.
gastric cancers (97). Furthermore, loss of FHIT protein expression was demonstrated immunohistochemically in the majority of gastric carcinomas (98). One somatic missense mutation was identified in exon 6 of the FHIT gene during a coding region analysis of 40 gastric carcinomas (99). Additional studies are needed to identify the critically altered targets on this chromosome, clarify the role FHIT plays in gastric tumorigenesis, and determine the role breakpoints in this region of 3p have in gastric cancer development.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


