Disease name
Aliases
Gene
Protein
Features
FIC1 deficiency
Byler disease, PFIC1
ATP8B1
FIC1
Multisystem disorder
Wide range of severity
BSEP deficiency
Byler syndrome, PFIC2
ABCB11
BSEP
Liver only affected
Spectrum of severity
TJP2 deficiency
–
TJP2
TJP2
Multisystem disorder
Wide range of severity
MDR3 deficiency
PFIC3
ABCB4
MDR3
Liver only affected
Wide range of severity
FIC1 Deficiency
Familial intrahepatic cholestasis protein 1 (FIC1) deficiency is an autosomal recessive condition with a wide phenotypic spectrum extending from mild recurrent to severe liver disease . FIC1 deficiency was initially called Byler’s disease, after the Amish family in which it was first described. Furthermore, the term Byler bile has been used as the description of coarsely granular canalicular bile seen in FIC1 deficiency on transmission electron microscopy.
It is characterized by hepatocellular cholestasis with low serum levels of γ-glutamyltransferase (GGT) activity and normal cholesterol levels. Presentation can be early on in life with recurrent cholestatic episodes and eventually progress to cirrhosis [1, 2]. Less severe disease can, however, present in adulthood. Presenting features consist of cholestasis, low serum albumin, epistaxis, and splenomegaly .
FIC1 is the protein encoded by the gene ATP8B1 [3–5], which is located at chromosome 18q21-q22. FIC1 (or ATP8B1) is a P-type ATPase, which is one of several aminophospholipid flippases present in different membranes. Its role in the liver, largely based on a mouse model, is in maintaining the gradient of phosphatidylethanolamine and phosphatidylserine towards the inner leaflet of the cell membrane. A transmembrane balance of lipids appears to be required in the presence of the high bile salt concentration in the canaliculus [6]. The normal canalicular membrane requires a high degree of detergent resistance in order to prevent damage from the detergent effect of bile itself. Paulusma et al. showed that by ex vivo infusion of bile acids in the knockout mouse, the extraction of aminophospholipids was greatly increased. In the human liver, several proteins are not present, or are in reduced quantities, in the canalicular membrane of patients with FIC1 deficiency. This observation still does not account for the cholestasis, as no transporter critical to bile formation has been identified to be absent. It appears more likely that these proteins are malfunctioning in an altered lipid environment of the canalicular membrane. As FIC1 deficiency is primarily characterized by cholestasis, studies have attributed this phenotype to a defective farnesoid X receptor (FXR) signalling pathway [7, 8]. Other groups, however, suggested that impaired FXR activity is secondary to cholestasis and not responsible for the phenotype [9] . The activity of FXR and its target genes remained uninterrupted in FIC1-depleted Caco-2 cells, created using small hairpin RNA and small interfering RNA, respectively, suggestive of an unimpaired FXR signalling mechanism in FIC1-deficient patients [10, 11]. In view of a proportion of phenotypically FIC1-like patients in whom no mutations were identified, a study looked into promoter and 5′ untranslated (5′-UTR) regions affecting gene regulation in human liver and small intestine tissue by 5′ rapid amplification of cDNA ends. Expression levels of ATP8B1 transcripts were determined by quantitative reverse-transcription polymerase chain reaction (qRT_PCR) and compared with the non-variable part of ATP8B1. Twelve different splicing variants and four novel untranslated exons located up to 71 kb upstream of the previously published exon 1 were identified in both tissue types. A number of transcription start sites were identified and the proximal promoter upstream of the major transcription start sites was also proven to be an essential regulatory element responsible for 70 % of total ATP8B1 transcriptional activity. In vitro, the main promoter was shown to drive constitutive ATP8B1 gene expression independent of bile acids [12] .
Altered gene expression demonstrated in extrahepatic sites may be contributory to the multisystemic nature of this cholestatic syndrome. Other disease features include diarrhea, malabsorption of fat-soluble vitamins , hearing loss, pancreatitis, renal tubular acidosis, delayed puberty , and growth failure. The multisystemic nature of the disease is supported by the widespread expression of the ATP8B1 gene including the liver, pancreas, kidney, and more widely in the small intestine [5].
Liver histology changes consist of cholestasis with bile plugs, periportal biliary metaplasia of hepatocytes in the absence of ductular proliferation in the portal tracks (Figs. 58.1, 58.2, and 58.3) .
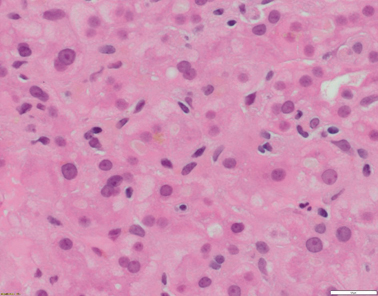
Fig. 58.1
Liver biopsy from a 20–year-old patient with ATP8B1 disease/FIC1. Hepatocytes are small and without anisocytosis. There is canalicular cholestasis. Of note, there is pale-staining bile within the canaliculi (H&E x400 magnification)
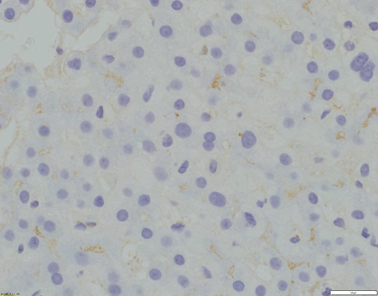
Fig. 58.2
Absence of canalicular GGT expression (x400 magnification)
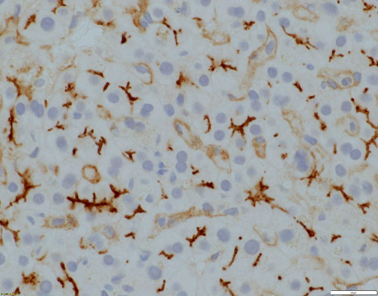
Fig. 58.3
Evident canalicular expression of CD10 (x400 magnification)
Early-onset patients, without treatment, usually progress to end-stage liver disease by early adulthood. First-line treatment includes antipruritic agents such as ursodeoxycholic acid (UDCA) [13], fat-soluble vitamin supplementation, and nutritional support. Failure of medical treatment would lead to first-line surgical treatment, which is currently partial external biliary diversion (PEBD), a technique that diverts an unknown proportion of bile externally. The consequences of this procedure certainly include interruption of the enterohepatic circulation of bile acids [14–17]. An external stoma can also be formed by using the appendix creating a cholecystoappendicectomy as reported by Rebhandl et al. [18]. Another alternative is ileal exclusion (IE), which has also been performed in cases with previous cholecystectomy or as rescue treatment [2, 15, 19, 20].
Liver transplantation (LT) is the next treatment option for these patients [16] but some extrahepatic symptoms will not improve, such as growth, and some may even become aggravated, such as diarrhea [14], liver steatosis, or kidney disease. Some of these patients may end up requiring multi-organ transplantation including liver, small bowel, kidneys, and pancreas (personal data) .
BSEP Deficiency
The major bile salt transporter of the canalicular membrane level is the BSEP , which is encoded by the gene ABCB11 [21] . The clinical condition associated with bile salt transport deficiency was previously known as “Byler’s syndrome”. The phenotype of BSEP-deficient patients can also vary from a mild benign recurrent intrahepatic cholestasis (BRIC) to a more severe type requiring LT [22–24]. ABCB11, expressed only in the liver, encodes BSEP; a member of the adenosine triphosphate (ATP)-binding cassette (ABC) family of transporters responsible for the transport of bile acids across the canalicular membrane. The locus for ABCB11 is at chromosome 2q24 with the most common European mutations being D482G (c.1445A > G) and E297G (c.890A > G) [21, 23]. BSEP deficiency leads to accumulation of bile salts within hepatocytes and has a subsequent effect on hepatocellular function. As anticipated, it has also been shown to have significant affinity for some of the main bile salts in human bile, such as glycocholate, taurocholate, and chenodeoxycholate [25] .
Severe BSEP deficiency presents within the first year of life and although variable, it is usually a non-relapsing severe progressive cholestasis and pruritus leading to fibrosis. Birth weight can be below normal range, especially in the non-D482G > E patients [22]. Serum bilirubin levels are not necessarily reflective of the degree of cholestasis as bilirubin is transported separately from bile salts. Like FIC1 deficiency, BSEP-deficient patients demonstrate low GGT activity but with a trend to higher cholesterol, significantly raised transaminases ( > threefold higher compared to FIC1 deficiency), alpha-fetoprotein , and serum bile acid concentrations. Other biochemical indices include fat-soluble vitamin deficiencies manifesting with coagulopathy and rickets. Gallstones have been reported in up to 32 % of patients [22]. Serum bile acid profiles demonstrate a high cholic acid to chenodeoxycholic acid ratio as reported in FIC1 patients. Reduced biliary bile salt concentration in BSEP patients is similar to that in progressive familial cholestasis (PFIC1) and in direct contrast to MDR3-deficient patients [3, 26, 27] .
Severe phenotypes have been associated with mutations leading to protein truncation or failure of protein production. In a series by Strautnieks et al., missense mutations were identified in 79 % of patients, many affecting protein processing and trafficking or protein structure [28]. Previous reports of ABCB11 missense mutations and single-nucleotide polymorphisms showed pre-messenger RNA (mRNA) splicing subsequently causing reduction in mRNA levels in a significant number of cases. These defects at the protein or mRNA level can have a detrimental impact in BSEP function [28].
Liver histology features consist of increased lobular inflammation and portal fibrosis, giant cell transformation of hepatocytes, and neonatal hepatitis with finely filamentous bile. Absence of liver immunohistochemistry for BSEP can assist in the diagnostic process [23, 29] (Fig. 58.4). Immunohistochemically detectable BSEP expression does not exclude functional BSEP deficiency [30]. In a previous series, 28 % of BSEP patients analysed exhibited some degree of BSEP staining and, in a small minority, expression was even considered normal [23] .
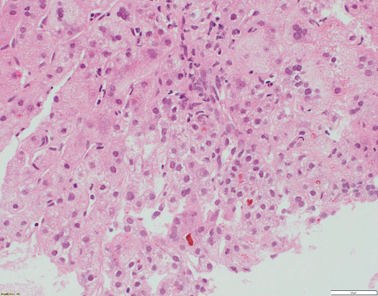
Fig. 58.4
Liver biopsy from a 7-month-old patient with ABCB11 disease/BSEP deficiency. There is bridging fibrosis and partial nodular transformation of the parenchyma (not shown). Hepatocytes show giant cell change and canalicular cholestasis. Lobular inflammation is present. BSEP immunostaining shows absence of canalicular expression, whilst canalicular GGT expression is preserved (not shown) (H&E stained slide at x200 magnification)
Hepatocellular carcinoma (HCC) or cholangiocarcinoma have been reported in a number of children on the background of severe neonatal hepatitis [31, 32] or proven BSEP deficiency [23, 32]. The risk appears to be higher in patients with two protein-truncating mutations (38 vs 10 %) compared to other genotypes. The exact mechanism of malignancy remains unclear, though further insight to the development of these tumours has been gained recently [33].
Treatment options for BSEP deficiency include antipruritic agents such as UDCA, rifampicin; liver enzyme inducers (phenobarbitone), nutritional support, and fat-soluble vitamin supplementation, where indicated. Partial external biliary diversion, ileal exclusion (IE) and LT are all recommended surgical treatment options [16, 20, 34, 35]. Hepatocellular malignancy remains an indication for early LT in BSEP patients. Patients with biallelic truncating mutations warrant close monitoring with liver ultrasonography and serum alpha-fetoprotein levels. Overall in terms of prognosis, patients with D482G mutation developed portal hypertension less frequently, developed fibrosis at an older age, and underwent LT at an older age as well [22] .
Recurrence of symptoms after LT has been described in BSEP-deficient patients, in contrast to other FIC syndromes. Jara et al. reported recurrence of cholestasis and pruritus following LT in BSEP patients with no evidence of cellular rejection on liver biopsy in 2009 [36]. In the same year, BSEP antibodies in the serum and at the hepatocyte canalicular membrane of a single patient who underwent two liver transplants were described [37]. The mechanism of action is thought to be that anti-BSEP antibodies form, which bind to an epitope in the intracanalicular domain of BSEP and subsequently block the function of the bile acid transporter. A multicentre series of six patients with recurrence of disease [38] was reported in 2010 . In all of these patients, treatment was extremely problematic, four underwent a repeat LT, and various management protocols were suggested. All reports have identified mutations (splice site, missense, truncating) leading to absence of BSEP protein expression on immunostaining. Modifications in immunosuppression, plasmapheresis, intravenous immunoglobulin courses, and single-pass albumin dialysis were used in isolation or jointly with limited effect in patients’ symptoms. Following the identification of BSEP antibodies in post-LT BSEP deficiency patients with cholestasis in the absence of rejection, an antibody-based treatment was suggested as potentially beneficial. Two cases of patients with BSEP deficiency following LT, had demonstrated evidence of functional BSEP deficiency treated successfully with two repeated 4-week courses of anti-cluster of differentiation CD20 monoclonal antibodies were subsequently reported [39, 40] .
TJP2 Deficiency
Since in one third of patients with normal GGT cholestasis mutations in either ABCB11 or ATP8B1 have not been identified [26], a cohort of 33 cholestatic children with relatively low-serum GGT levels were studied recently [41] . Twelve patients from eight consanguineous families with novel protein truncating mutations in the TJP2 gene were identified. The phenotype of these patients consists of early presentation within the first couple of months of life with low-serum GGT and nine of them underwent LT by their first decade of life. Patients can develop portal hypertension, persistent pruritus, and malabsorption . A single patient died at 13 months. Liver immunohistochemical findings consist of lack of TJP2, reduced staining of Claudin-1 (CLDN1) with normal distribution of Claudin-2 (CLDN2), both proteins essential to cellular tight junctions (Figs. 58.5, 58.6, and 58.7). On transmission electron microscopy, elongation of the tight junctions with sparsity of the zona occludens (ZO) is also seen. Extrahepatic involvement is present in some cases and consists of chronic respiratory disease or neurological complications such as subdural haematomas. Mutations in the TJP2 gene have been previously associated with familial hypercholanaemia (FHC), a non progressive cholestatic disorder, described in the Amish population [42]. In that report, out of all 17 individuals with FHC, screened 11 patients in eight families were found homozygous for an incompletely penetrant missense mutation in TJP2, with alterations in the cellular bile acid concentration gradient .
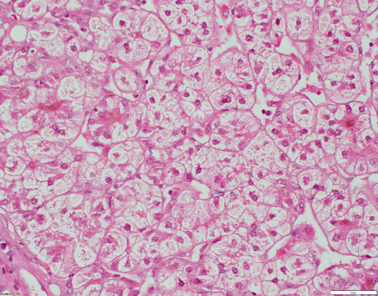
Fig. 58.5
Liver biopsy from a 4-year-old patient with TJP2 disease. There is extensive bridging fibrosis (not shown). Hepatocytes are oedematous and show rosetting. Canalicular cholestasis is present (H&E x200 magnification)
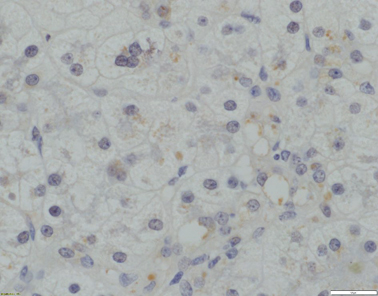
Fig. 58.6
Absence of canalicular TJP2 expression (x400 magnification)
Only gold members can continue reading. Log In or Register to continue
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


