CHAPTER 32 Endocrine Tumors of the Pancreas and Gastrointestinal Tract
GENERAL CONSIDERATIONS
HISTORICAL ASPECTS
In 1927,1 five years after the discovery of insulin, the first pancreatic hormone-producing tumor syndrome was described in a patient with a metastatic islet cell tumor and hypoglycemia, and whose tumor extracts had hypoglycemic effects. Numerous other pancreatic endocrine tumors (PETs) have since been described (Table 32-1), with the description of Zollinger-Ellison syndrome in 1955,2 the Verner-Morrison syndrome caused by a diarrheogenic-producing tumor in 1958,3 glucagonoma syndrome by Mallinson in 1974,4 the somatostatinoma syndrome in 1977,5,6 and GRFomas (pancreatic tumors secreting growth hormone-releasing factor) in 1982.7,8 PETs secreting adrenocorticotropic hormone (ACTH; ACTHomas) are also included because 4% to 16% of cases of ectopic Cushing’s syndrome are caused by ACTH-secreting PETs.9–11 PETs causing the carcinoid syndrome (see Chapter 31),12 tumors secreting renin and causing hypertension,13 luteinizing hormone (LH) resulting in masculinization or changes in libido,14 erythropoietin resulting in polycythemia,15 and parathyroid hormone-related protein (PTHrP), resulting in hypercalcemia,16 have also been described. PETs secreting calcitonin17 are proposed to cause a distinct syndrome with diarrhea. However, too few cases have been well described to include this syndrome. Furthermore, other causes of hypercalcitonemia, such as medullary thyroid cancer, are only associated with diarrhea in 25% to 42% of patients.18 Other functional hormonal syndromes have been described, with nonpancreatic, primarily intra-abdominal neuroendocrine and non-neuroendocrine tumors, including secretion of GLP-2 (glucagon-like peptide-2), causing intestinal villous hypertrophy (enteroglucagonomas) and secretion of GLP-1 causing hypoglycemia and delayed GI transit,19 and intestinal and ovarian tumors secreting peptide YY resulting in altered intestinal motility and constipation.20 PETs secreting ghrelin have been described21 but were not associated with acromegaly, increased serum growth hormone levels, or increased insulin growth factor-1 (IGF-1) concentrations.
PETs are classified as functional if associated with a clinical syndrome caused by hormone release by the tumor or nonfunctional if not associated with a clinical syndrome caused by hormone release (see Table 32-1). In the nonfunctional category are included nonfunctional PETs (NF-PETS), which have the histologic characteristics of a PET but no associated elevation in plasma hormone levels or clinical syndrome, as well as PETs that release pancreatic polypeptide (PPomas), ghrelin, neurotensin (neurotensinomas), or other peptides that do not cause a distinct clinical syndrome.9,22,23
PREVALENCE AND INCIDENCE
PETs account for 1% to 10% of tumors arising in the pancreas.22,24,25 The overall prevalence of functional PETs is low, reported to be approximately 10/million (1/100,000). In contrast, the prevalence of PETs in autopsy studies is higher, 0.5% to 1.5%.26 The annual incidence of PETs is reported at 1 to 4 cases/million/year.9 Nonfunctional PETs account for 14% to 30% of all PETs in most studies, but they are as high as 60% to 80% in some studies.27 Insulinomas and gastrinomas occur with an equal annual incidence of 0.5 to 3 cases/million.28–30 VIPomas are 12.5% as common and glucagonomas 6% as common as insulinomas and gastrinomas. Somatostatinomas are very rare31,32 and the incidence of GRFomas, PETs secreting renin, erythropoietin, or LH or PETs causing hypercalcemia is unknown (see Table 32-1).
ORIGIN AND HISTOLOGIC FEATURES
PETs are often called islet cell tumors but it is unproven that they originate from the pancreatic islets.9,33,34 These tumors frequently contain ductular structures, produce hormones not normally present in the adult pancreas, such as gastrin and vasoactive intestinal peptide (VIP), and may produce multiple hormones.35 It has been suggested that these tumors represent a dedifferentiation of an immature stem cell. The finding of the ductular structures in many PETs and the budding off of endocrine cells from ductules during ontogenesis of the pancreas has led to the suggestion these tumors are ductular in origin.36
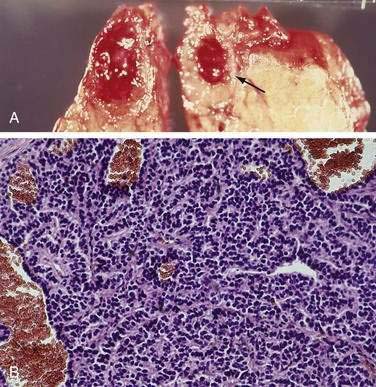
It was originally proposed that PETs might originate from cells that are part of the diffuse neuroendocrine cell system.9,34,37,38 These cells share certain cytochemical properties (amine precursor uptake and decarboxylation) and the tumors have been called APUDomas.33 Ultrastructurally, the cells often have electron-dense granules and produce multiple regulatory hormones and amines, neuron-specific enolase, synaptophysin, and chromogranin A or C. These cells are thought to give rise to carcinoid tumors, medullary carcinomas of the thyroid, melanomas, and pheochromocytomas, and there are marked similarities in the histology of these tumors and PETs. Histologically, PETs consist of a relatively homogeneous sheet of small round cells with uniform nuclei and cytoplasm (Fig. 32-1). Mitotic figures are uncommon.33 Malignancy can be determined only by metastases or invasion and cannot be predicted by light microscopic or ultrastructural studies.20,28 At present, controversy still surrounds the exact cell of origin for PETs, with some recent studies suggesting they originate from islet cells and others supporting a ductular origin.34,36
Most PETs produce multiple gastrointestinal hormones, which can be localized by immunocytochemical methods.9,33 In many studies, most functional and nonfunctional PETs had cells immunoreactive to peptides that were not causing clinical symptoms.35 It is unclear why usually only one or no clinical syndrome is seen, despite the immunochemical occurrence of multiple hormones.28 A functional PET syndrome should be diagnosed only if the appropriate clinical symptoms are present, not only based on immunocytochemistry.
PETs frequently produce chromogranins or the alpha or beta subunit of human chorionic gonadotropin (HCG), which can be localized by immunocytochemistry or by documenting elevated circulating levels.9,28,33 Chromogranins are water-soluble acidic glycoproteins that are present in almost all endocrine or neuronal tissues.26 Plasma chromogranin A levels are elevated in more than 90% of patients with various PETs and carcinoid tumors (see Chapter 31). Although some have suggested that elevations of the alpha or beta subunit of HCG or of chromogranin A may be indicative of malignancy, this is not established.9,28 Furthermore, some studies have reported that serial measurements of chromogranin levels may be useful for monitoring tumor growth39–42 or as a marker of survival.43
In an occasional patient, a second clinical hormone tumor syndrome may be present initially or develop with time.9,28 Whereas one study reported that this occurred in 7% of all patients with PETs during a three-year follow-up,9 another study35 has reported that this is a rare occurrence, occurring at a rate of 2/100 followed over ten years. However, there appears to be a high incidence of the development of Cushing’s syndrome in patients with a functional PET, especially in those with gastrinomas.44,45
CLASSIFICATION
PETs are classified clinically according to the functional syndrome produced (see Table 32-1). Although clinical syndromes have been attributed in some studies to patients with PETs who had elevated plasma levels of neurotensin or pancreatic polypeptide (PP), the existence of these syndromes has not been established with certainty.9,35,46 PETs can be associated with four different inherited disorders: multiple endocrine neoplasia type I (MEN-I), von Hippel-Lindau disease, tuberous sclerosis, and neurofibromatosis-1 (von Recklinghausen’s disease).47–49 These association are important to recognize because family screening may be needed and because these PETs may have a different natural history (see later).28
It has been proposed9,33 that the terms PET and carcinoid tumor be replaced by the term neuroendocrine tumor (NET) and a new classification based on clinical and morphologic categories be used (see Chapter 31). This WHO classification classifies all NETs into well-differentiated endocrine tumors or carcinomas, poorly differentiated endocrine tumors, or mixed exocrine-endocrine tumors, which better allows comparisons of NETs from the pancreas and other gastrointestinal sites. The well-differentiated NETs of the pancreas are specifically divided into well-differentiated tumors and well-differentiated endocrine carcinomas. The well-differentiated endocrine tumors are further divided into those with benign behavior (confined to pancreas, nonaggressive, <2 cm in size, functional or nonfunctional), and uncertain behavior (confined to pancreas, >2 cm in size, or with angioinvasion, functional or nonfunctional). The well-differentiated endocrine carcinomas of the pancreas show low-grade malignancy, with gross invasion and/or metastases and can be functional or nonfunctional. In this chapter, the term PET will be retained because of its widespread use. Recently, for the first time, a TNM classification has been proposed for PETs and for other GI neuroendocrine tumors (see Table 31-1).50 This TNM classification is based on the level of tumor invasion, tumor size, tumor extent, and with grading using the mitotic index or the proliferative index, Ki-67. The importance of these classification systems for predicting prognosis will be discussed in a later section.
PATHOPHYSIOLOGY
In various series, in a small percentage of the patients with a functional syndrome in whom no tumor was found preoperatively and at the time of surgery, hyperplasia of the pancreatic islets was regarded as a possible cause of the disease.9,28 Beta cell hyperplasia or nesidioblastosis, which is a subtype of beta cell hyperplasia consisting of the proliferation of islet cells from pancreatic ducts, is reported to be a cause of hypoglycemia and hyperinsulinemia in a number of infants and newborns. Recently, this condition has been recognized in adolescents and adults and occurs in 5% of patients with hyperinsulinism.51,52 It has been suggested that Zollinger-Ellison syndrome and Verner-Morrison syndrome are caused in up to 10% of cases by hyperplasia of pancreatic ducts, producing gastrin and VIP, respectively. However, this concept has not been substantiated by immunocytochemical studies and thus is not generally accepted.
MOLECULAR PATHOGENESIS
Until recently, the molecular pathogenesis of neuroendocrine tumors (NETs) (carcinoids and PETs) was largely unknown.53,54 Numerous studies have demonstrated that in contrast to most common nonendocrine tumors (e.g., colonic or pancreatic adenocarcinoma), mutations in common oncogenes (e.g., ras, fos, myc, src, jun) and common tumor suppressor genes (e.g., p53, retinoblastoma gene) are uncommon in most NETs (carcinoids, PETs).26,38,55 Recent studies have provide evidence that alterations in the MEN-I gene, p16/MTS1 tumor suppressor gene, DPC4/Smad 4 gene, amplification of the HER-2/neu proto-oncogene, increased expression of growth factors and/or their receptors (endothelial growth factor [EGF], hepatocyte growth factor, platelet-drived growth factor [PDGF]), and deletions of a possible unknown tumor suppressor gene on chromosome 1 or 3p may all be important.26,38,53–57 Alterations in the MEN-I gene occur in up to one third of sporadic (i.e., noninherited) PETs58 and alterations in the p16/MTS1 gene occur in 50% to 92% of PETs and thus may be particularly important. Alterations in the MEN-I gene are discussed in the next section. Genomic-wide allelotyping and comparative genomic hybridization studies have demonstrated that chromosomal losses (especially on 1p, 3p, 3q, 6q, 9q, 12q) and chromosomal gains (especially in 7q, 17q, 17p, 20q) frequently occur in PETs and carcinoids, but their frequency varies markedly in these two gastrointestinal (GI) neuroendocrine tumors, providing evidence that they have a different molecular pathogenesis. Gene expression profiling using microarrays has identified a large number of genes that are altered in PETs in comparison to normal islets or between PETS with different degrees of aggressiveness.26,59–62 At present, it is not clear which of the different genes altered in the different studies will have important prognostic implications or provide important insights into their pathogenesis that will lead to new treatments.
MULTIPLE ENDOCRINE NEOPLASIA
There are three well-established MEN syndromes that can be distinguished by the presence or absence of PETs; medullary thyroid carcinoma, parathyroid disease, pheochromocytoma and a specific phenotype.47,49,63 Each of these syndromes—MEN type I, MEN type IIa, and MEN type IIb—has autosomal dominant inheritance. MEN-I, or Wermer’s syndrome, is considered in detail later; it is characterized by hyperparathyroidism and PETs without the presence of medullary thyroid carcinoma, pheochromocytoma, or unusual phenotype. MEN-IIa, or Sipple’s syndrome, is characterized by bilateral medullary thyroid carcinoma and pheochromocytomas (in 20% to 40%); when they occur, they are bilateral in 70%, with hyperparathyroidism in 17% but without the occurrence of PETs or a specific phenotype. MEN-IIb includes bilateral medullary thyroid carcinoma, which often appears at an early age and appears to be more aggressive than these tumors in patients with MEN-IIa. Pheochromocytomas, when they occur, are bilateral in 70%. Parathyroid disease is seldom present in MEN-IIb, and patients have a characteristic phenotype, with multiple mucosal neuromas, frequently marfanoid habitus, puffy lips, prominent jaw, pes cavus, and medullated corneal nerves, but no PETs. The genetic defect in MEN-I is located on the long arm of chromosome 11 and is caused by mutations in a 10-exon gene encoding for a 610–amino acid protein, MENIN, a nuclear protein that interacts with the AP1 transcription factor, Jun D, nuclear factor κB (NF-κβ), pem, SMAD3, RPA2 (a DNA processing factor), FAN CD2 (a DNA repair factor), nucleoside diphosphate kinase, NM23β, and various cytoskeleton-associated proteins.64
The development of MEN-I endocrine tumors conformed to Knudsen’s9 two-hit model theory of neoplasm, with an inherited mutation in one chromosome unmasked by a somatic deletion or mutation of the other normal chromosome, thereby removing the suppressor effect of the normal gene. In PETs from patients without MEN-I, up to 90% have loss of heterozygosity on chromosome 11 and 27 % to 39% have mutations in the MEN-I gene.26,53,54,58 This suggests that sporadic PETs share a similar tumorigenesis to PETs that occur in patients with MEN-I, which principally involves deletion of a tumor suppressor gene. The MEN-II syndromes are caused by alterations in the pericentromeric region of chromosome 10 in the RET proto-oncogene, which is a 21-exon gene encoding for a tyrosine kinase receptor. Mutations in a cysteine-rich extracellular portion of the receptor primarily cause MEN-IIa, whereas mutations in the gene region encoding the intracellular catalytic core of the tyrosine kinase domain cause MEN-IIb.
In patients with MEN-I, hyperparathyroidism is the most common clinical abnormality, occurring in 78% to 97% (Table 32-2).9,28,47–49 Functional PETs are the second most common clinical abnormality, occurring in 81% to 82% of patients. Gastrinomas occur in 54%, whereas insulinomas, glucagonomas, and VIPomas occur in 18%, 3%, and 3% of patients, respectively.65 Nonfunctional PETs and PPomas may be the most common PET in patients with MEN-I because they are almost always found in histologic studies.66 However, large nonfunctional PETs causing symptoms occur in only 0% to 13% in various series. Many patients without a functional PET do not routinely undergo surgical exploration,56,67,68 and imaging studies routinely miss most small PETs smaller than 1 cm.26,69 Therefore, the true incidence of asymptomatic PETs in these patients is unknown.
Table 32-2 Frequency of Features of Patients with Multiple Endocrine Neoplasia Type I
| FEATURE | AVERAGE FREQUENCY (RANGE), % OF ALL PATIENTS |
|---|---|
| Hyperparathyroidism | 97 (78-100) |
| Pancreatic endocrine tumors | 81-82 |
| Nonfunctional or PPomas | 80-100 (microscopic), 0-13 (symptomatic) |
| Gastrinomas | 54 (20-61) |
| Insulinomas | 18 (7-31) |
| Glucagonomas | 3 (1-6) |
| VIPomas | 1 (1-12) |
| Somatostatinomas | 0-1 |
| GRFoma | <1 |
| Pituitary tumors | 54-65 (15-100) |
| Prolactin-secreting | 15-46 |
| Growth-hormone secreting | 6-20 |
| Cushing’s syndrome | 16 |
| Adrenal tumors | 27-36 (symptomatic, <2%) |
| Cortical adenomas | |
| Hyperplasia, carcinoma (uncommon) | |
| Carcinoid tumors | |
| Gastric (ECLoma) | 7-35 (symptomatic, <5%) |
| Lung | 0-8 |
| Thymic | 0-8 |
| Skin tumors | 40-100 (angiofibromas) |
| Angiofibromas > collagenoma > café-au-lait macules > lipomas | 88, 72, 38, 34 (symptomatic, <1%) |
| CNS tumors—meningiomas, ependymomas, schwannomas | 0-8, 0-1 (symptomatic, <1%) |
| Smooth muscle tumors—leiomyomas, leiomyosarcomas | 1-7 (symptomatic, <1%) |
| Thyroid tumors—adenomas | 0-10 (0-30; symptomatic, <1%) |
CNS, central nervous system; ECL, enterochromaffin-like cell; GRF, growth hormone-releasing factor; PP, pancreatic polypeptide; VIP, vasoactive intestinal peptide.
From references 28, 46–49, and 65.
Pathology studies9,47,48,66 have demonstrated that in almost every patient with MEN-I, the pancreas demonstrates diffuse microadenomatosis, with or without larger tumors. With immunocytochemistry, PP is most frequently seen followed by glucagon and insulin, with gastrin rarely found. These results are consistent with clinical studies that have demonstrated that gastrinomas in more than 80% of patients with MEN-I and Zollinger-Ellison syndrome are located in the duodenum.28,67,68,70,71
MEN-I is present in 20% to 25% of patients with gastrinomas, 4% of patients with insulinomas, 13% to 17% of patients with glucagonomas, 33% of patients with GRFomas, 9% of patients with VIPomas, and 7% with somatostatinomas.9,27,28,47,48,72–78 Characteristically, hyperparathyroidism is the initial manifestation of MEN-I, usually presenting in the third decade of life, followed by the development of a PET in the fourth to fifth decade.49 It is important to recognize whether a patient has MEN-I because patients with and without MEN-I differ in their clinical presentation, in the possibility of surgical cure, and in the clinical and diagnostic approach to the tumor.56,67,68 Patients with MEN-I may develop more than one PET over time so long-term follow-up will differ from that of a patient without MEN-I. Screening of other family members will be indicated in patients with MEN-I, whereas it will not be in patients with sporadic disease. In some PETs, the presence of the hypercalcemia caused by the hyperparathyroidism may affect release of the hormones by the tumor.79
OTHER INHERITED SYNDROMES ASSOCIATED WITH PANCREATIC ENDOCRINE TUMORS
Three inherited phacomatoses have an increased occurrence of PETs—von Hippel-Lindau disease (VHL), von Recklinghausen’s disease (neurofibromatosis-1 [NF-1]), and Bourneville’s disease (tuberous sclerosis).26,46,47 VHL is caused by a defect on chromosome 3p25 encoding for a 232–amino acid protein, pVHL, that forms a complex with a number of proteins, including elongin B and C as well as Cullin 2, which regulate ubiquitin-dependent proteolysis of large cell proteins.80 VHL mutations result in altered transcriptional regulation, resulting in pathologic changes in angiogenic, growth, and mitogenic factors. In 10% to 17% of patients with VHL, a PET is seen which is usually asymptomatic and nonfunctional (>98%), although occasional insulinomas and VIPomas are described. The mean age at diagnosis of a PET in VHL is 29 to 38 years. Most patients have a single PET (67% to 70%). Malignant PETs occurs in 8% to 50% of VHL patients with PETs, with liver metastases in 9% to 37%.
NF-1 is caused by a defect on chromosome 17q11.2 encoding for a 2845–amino acid protein, neurofibromin, which functions as a ras signaling cascade inhibitor.9,81 In various series, from 0% to 10% of NF-1 patients develop a carcinoid tumor, usually in the periampullary region (54%) of the duodenum (see Chapter 31).78,82 Most of these duodenal carcinoids are somatostatinomas by immunocytochemistry, but they rarely produce the somatostatinoma syndrome.47 NF-1 has rarely been associated with Zollinger-Ellison syndrome and insulinomas.46 NF-1 accounts for 48% of all duodenal somatostatinomas and approximately 25% of all ampullary carcinoid tumors. These tumors frequently (63%) show psammoma bodies histologically, and metastases to liver and or lymph nodes occur in 30%.
Tuberous sclerosis is caused by mutations in the 1164–amino acid protein, hamartin (TSC-1), or the 1807–amino acid protein, tuberin (TSC-2).47,83 These two proteins are important in regulating the PI3K signaling cascade (see Chapter 3) and also for regulation of the small GTPase, Rheb, which play important roles in the regulation of protein translation and synthesis, growth, and proliferation, as well as maintenance of cellular energy levels. A few cases of nonfunctional and functional PETs (insulinomas and gastrinomas) have been reported in patients with tuberous sclerosis.26,46
INSULINOMAS
DEFINITION
Insulinomas are insulin-secreting tumors that primarily originate in the pancreas and cause symptoms as a result of hypoglycemia (Table 32-3).
Table 32-3 Frequency of Symptoms and Signs in Patients with Insulinoma
| FINDING | FREQUENCY (%) |
|---|---|
| Any Time During Clinical Course | |
| Neuropsychiatric symptoms (loss of consciousness, confusion, dizziness, diplopia) | 92 |
| Confusion or abnormal behavior | 80 |
| Obesity | 52 |
| Amnesia or coma | 47 |
| Cardiovascular symptoms, palpitations, tachycardia | 17 |
| Convulsions (grand mal) | 12 |
| Gastrointestinal symptoms (hunger, vomiting, pain) | 9 |
| During First Attack | |
| Neuroglycopenic symptoms | |
| Visual disturbances (diplopia, blurred vision) | 59 |
| Confusion | 51 |
| Altered consciousness | 38 |
| Weakness | 32 |
| Transient motor defects, hemiplegia | 29 |
| Dizziness | 28 |
| Fatigue | 27 |
| Inappropriate behavior | 27 |
| Speech difficulty | 24 |
| Headache | 23 |
| Seizure | 23 |
| Syncope | 21 |
| Difficulty concentrating or thinking | 19 |
| Paresthesias | 17 |
| Memory loss | 15 |
| Lethargy | 12 |
| Stupor | 12 |
| Amnesia | 8 |
| Ataxia | 4 |
| Disorientation | 4 |
| Mental change | 4 |
| Adrenergic symptoms | |
| Sweating | 43 |
| Tremulousness | 23 |
| Hunger, nausea | 12 |
| Palpitations | 10 |
PATHOPHYSIOLOGY AND PATHOLOGY
Insulinomas almost always (98.2%) occur in or are attached to the pancreas.9,26,29,30 An occasional insulinoma presenting as a carcinoid tumor has been reported in the duodenum, ileum, and lung, but truly ectopic insulinomas are rare (1% to 3%).84 Insulinomas are evenly distributed in the pancreas, with approximately one third in the pancreatic head, body, and tail.9,26,84–86 Insulinomas are usually small. In one large series, 5% were less than 0.5 cm, 34% were 0.5 to 1 cm, 53% were 1 to 5 cm, and only 8% were more than 5 cm.85
Insulinomas are usually solitary, with multiple tumors occurring in only 2% to 13% of cases.9,26,29,30 If multiple insulinomas are found, MEN-I should be suspected.47 Insulinomas are generally well encapsulated, firmer than normal pancreas, and highly vascular. Only 5% to 16% of insulinomas are malignant.87 Malignant tumors are generally larger, averaging 6 cm in one series, and 5% of patients have metastases at presentation.88 Metastases are usually to the liver (47%), regional lymph nodes (30%), or both.
Among adults with hyperinsulinism and pancreatic islet cell disease, histologic studies have shown a solitary insulinoma in 86% of cases, adenomatosis in 5% to 15%, nesidioblastosis in 4%, and islet hyperplasia in 1%.87 Adenomatosis consists of multiple macroadenomas or microadenomas and occurs especially in patients with MEN-I. A second diffuse lesion is nesioblastosis, a condition in which islet cells bud off from ductular structures and are mixed with lobular elements. This condition previously was reported almost exclusively in infants and children, but has been recognized in 5% of adults and adolescents with hyperinsulinism.9,51,89 Diffuse islet cell hyperplasia, which consists of excessive and diffuse proliferation of beta cells in the islets, has been reported in adults.85 Of 1137 cases of organic hyperinsulinism, only 6% had diffuse islet cell hyperplasia and another 0.6% had both an insulinoma and diffuse islet cell disease. At present, it is unclear whether many of these cases were in fact nesioblastosis because in most cases appropriate immunofluorescence staining methods were not applied.
Insulin is synthesized and stored in beta cells of the pancreatic islets.29,30 Insulin is synthesized in the rough endoplasmic reticulum as preproinsulin, from which proinsulin is liberated and transferred to the Golgi of the cell.90 Proinsulin consists of a 21–amino acid alpha chain and a 30–amino acid beta chain connected by a 33–amino acid connecting peptide (C-peptide). In secretory granules, a protease excises the C-peptide and thus, when secretion occurs, the C-peptide and the double-stranded insulin molecule are released in equimolar amounts. Small amounts of intact proinsulin remain in granules and are also released; this can be detected in the plasma. Proinsulin contains the alpha and beta chains of insulin and, because most insulin antibodies used in radioimmunoassays recognize moieties on these chains, they also recognize proinsulin. Normal subjects have less than 25% of their total serum insulin as proinsulin, whereas over 90% of patients with insulinomas have an elevated proportion of proinsulin relative to total insulin.91
CLINICAL FEATURES
Insulinomas can occur at any age but are rare in adolescents, usually occurring in patients between 20 to 75 years. A large majority occur between the ages of 40 to 45 years and 60% of patients are women.9,26,29,30 Symptoms are caused by hypoglycemia (see Table 32-3) characteristically associated with fasting and thus more frequently occur when a meal is delayed, missed, or before breakfast. Symptoms may also occur during exercise. In one study,87 26% of patients had symptoms during or after an overnight fast, 27% had symptoms prior to lunch or dinner, 8% had symptoms only after a missed meal, 29% had symptoms only before lunch or dinner, and only 9% were uncertain about when their symptoms occurred. The hypoglycemia with fasting or exercise, which is characteristic of insulinomas, differs temporally from hypoglycemia, which occurs after meals (postprandial hypoglycemia). Postprandial hypoglycemia can be caused by a number of other unrelated conditions and is increasingly being reported in patients after various gastric bypass surgery procedures for obesity.92 Most symptoms of insulinomas (82% to 92% of patients)85,93 are caused by neuroglycopenia, because glucose is the main source of energy for the brain. Neuroglycopenic symptoms include somnolence, visual disturbances, irritability, abnormal behavior, confusion, amnesia, paresthesias, stupor, drowsiness, coma, and seizures. Symptoms of hypoglycemia can also be caused by catecholamine release (adrenergic symptoms) and include anxiety, palpitations, weakness, fatigue, headache, tremor, and sweating. Coma occurs in up to 53% of patients and convulsions in 12%. In one study of symptoms of a first attack, 49% of patients initially had both neuroglycopenic and adrenergic symptoms, 38% had neuroglycopenic symptoms only, 12% had adrenergic symptoms only, and 1% had no symptoms. Of the neuroglycopenic symptoms, visual disturbances (57%), confusion (51%), and altered consciousness (38%) are the most common. Of the adrenergic symptoms, sweating (43%) and tremulousness (23%) are the most common. Patients frequently learn to avoid symptoms by eating frequently and obesity may result. In one study,87 40% of patients with organic hypoglycemic were overweight. The average duration of neuroglycopenic symptoms prior to diagnosis is often prolonged, being more than three years in 25% of patients and more than five years in 20%.
DIAGNOSIS AND DIFFERENTIAL DIAGNOSIS
The key to establishing the diagnosis of insulinoma is suspecting by clinical history—that the symptoms could be caused by hypoglycemia—and establishing the relationship of the symptoms to fasting.9,29,30,89 Whipple’s triad, published in 1938, and long used as diagnostic criteria for insulinoma, was based on this association, consisting of characteristic hypoglycemia symptoms, the presence of hypoglycemia (blood sugar < 50 mg/dL), and relief of symptoms following glucose ingestion. Unfortunately, these symptoms are not specific for insulinoma.87
Organic hypoglycemia is generally defined as a fasting blood glucose level lower than 40 mg/dL. In healthy individuals, after an overnight fast, plasma glucose values usually do not decrease below 70 mg/dL.87 After an overnight fast, only 53% of patients with insulinoma have a blood glucose level below 60 mg/dL and only 39% below 50 mg/dL. However, if a blood glucose determination is combined with a concomitant plasma insulin level, this insulin level will be inappropriately elevated in 65% of patients. Hypoglycemia can be classified as a fasting hypoglycemia or postprandial (reactive) hypoglycemia, of which there are a number of different causes (Table 32-4






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


