Effects of Chronic Kidney Disease on Metabolism and Hormonal Function
Jonathan Himmelfarb
Ian de Boer
Bryan Kestenbaum
INTRODUCTION
In health, blood is filtered continuously by the kidneys, resulting in near complete homeostasis in the chemical composition of the blood compartment over a wide range of nutrient and fluid intakes. Kidney function thus plays a critical role in maintaining circulatory and organ system functional homeostasis, and the loss of kidney function in chronic kidney disease (CKD) and especially end-stage renal disease (ESRD) leads to dysregulation of many metabolic pathways. This results clinically in the uremic syndrome, characterized by subtle dysfunction of many organ systems.
The kidney also regulates body homeostasis not only by excretory function but by several synthetic and degradative properties dependent on glomerular and tubular epithelial cell function. These properties include synthesis of hormones, degradation of peptides and low-molecular-mass proteins (less than 50 kilodaltons [kDa]), and metabolic events aimed at conserving energy and regulating the composition of body fluids. The kidney is the site of synthesis of a number of hormones (i.e., erythropoietin [EPO], 1,25-dihydroxyvitamin D3 [1,25-dihydroxycholecalciferol], and renin) and is an important catabolic site for several polypeptide hormones (e.g., insulin, glucagon, and parathyroid hormone [PTH]) and glycoproteins (Table 2-1).
Solute Accumulation
In CKD, as glomerular filtration rate (GFR) decreases solutes that are excreted by the kidney (such as creatinine and urea) accumulate in body fluids, and the plasma concentration of these solutes increases. Other solutes, including phosphates, sulfates, uric acid, and hydrogen ions, also can accumulate in body fluids. Accumulation of hydrogen ions is because of impairment of both ammoniagenesis and hydrogen ion secretion in tubular cells, resulting in the development of metabolic acidosis. Although the most profound changes occur with severe impairment of GFR, many of these abnormalities with associated adaptive or maladaptive responses begin at GFR of 60 mL/min/1.73 m2 or more. As CKD progresses, other compounds that are retained in body fluids include phenols, guanidines, organic acids, indols, myoinositol and other polyols, polyamines, β2-microglobulin, certain peptides, urofuremic acids, and trace elements, such as aluminum, zinc, copper, and iron. The accumulation of these toxic substances can lead to hormonal deficiencies (testosterone, fetuin, etc.), inability to appropriately respond to stimuli (insulin resistance, EPO resistance, etc.), or overproduction (prolactin). As kidney function declines, patients’ abilities to adapt to changes in
dietary intake, particularly those involving sodium, potassium, phosphorus, and water, become more restricted. Solute and water excretion per nephron increases as kidney function decreases, but this is counterbalanced by the fewer number of functional nephrons. As kidney disease progresses, the capacity to respond to changes in the intake of sodium, other solutes, and water becomes less flexible, and eventually this loss of capacity can result in changes in the volume and composition of the extracellular fluid. Thus, dietary intake in patients with CKD often needs to be adjusted.
dietary intake, particularly those involving sodium, potassium, phosphorus, and water, become more restricted. Solute and water excretion per nephron increases as kidney function decreases, but this is counterbalanced by the fewer number of functional nephrons. As kidney disease progresses, the capacity to respond to changes in the intake of sodium, other solutes, and water becomes less flexible, and eventually this loss of capacity can result in changes in the volume and composition of the extracellular fluid. Thus, dietary intake in patients with CKD often needs to be adjusted.
Table 2-1. Components of kidney function | |||||||||
|---|---|---|---|---|---|---|---|---|---|
|
METABOLIC PATHWAYS FREQUENTLY ALTERED IN CHRONIC KIDNEY DISEASE
Chronic kidney disease is known to be associated with alterations in multiple metabolic pathways. Recent studies indicate that these alterations might lead to a milieu highly conducive to development of atherosclerosis and might explain, at least to some extent, the exaggerated cardiovascular disease risk in this patient population (Fig. 2-1).
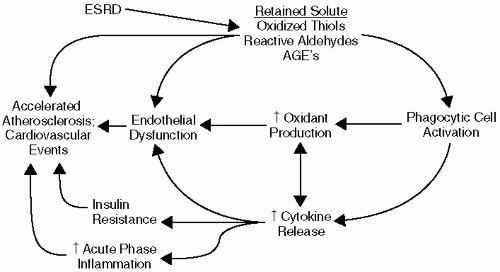 Figure 2-1. A proposed mechanism for uremia-induced CVD risk. |
Inflammation
Acute-phase inflammation is a common feature of CKD and ESRD. It has been demonstrated that 30% to 50% of advanced patients with CKD have serologic evidence of an activated inflammatory response in cross-sectional studies. In longitudinal studies virtually all patients undergoing dialysis have evidence of intermittent inflammation. Elevated plasma levels of the proinflammatory cytokine interleukin 6 (IL-6) and the prototypical acute-phase protein C-reactive protein (CRP) have emerged as powerful independent predictors of cardiovascular events and mortality, often exceeding the prognostic value of traditional cardiovascular risk factors such as low-density lipoprotein (LDL) cholesterol. Moreover, plasma IL-6 and CRP levels do not change significantly over the course of time after initiation of dialysis therapy, suggesting that dialysis does not completely improve the inflammatory metabolic alterations associated with loss of kidney function. A plethora of additional inflammatory markers have been studied in kidney disease, including sICAM-1, Serum Amyloid A, IL-8, IL-18, myeloperoxidase, sCD40 ligand, and matrix metalloproteinase-9. Fibrinogen is also an important acute-phase reactant, and fibrinogen levels significantly predict cardiovascular events in patients with CKD undergoing peritoneal and hemodialysis.
The underlying etiology of the augmented inflammatory response in kidney disease remains poorly understood. The hemodialysis procedure itself contributes to a proinflammatory milieu by increasing the synthesis of fibrinogen, IL-6, and other proinflammatory peptides. This is primarily from bioincompatibility because of membrane and dialysate exposure during the extracorporeal procedure. Subclinical vascular access infections may also contribute to augmented inflammation in many patients undergoing dialysis. However, while multiple dialysis-related factors can contribute to maintenance of a chronic inflammatory response, recent data demonstrating that plasma IL-6 and CRP levels are already elevated in earlier stages of CKD suggest a more proximate relationship to loss of kidney function. Loss of kidney function is directly associated with reduced renal clearance of cytokines, complement peptides, oxidants, and other proinflammatory solutes. Biological priming of inflammatory cells may also be an important contributor to increased inflammation in CKD. Inflammation is exacerbated by comorbid conditions such as diabetes mellitus and inflammatory renal disease.
Oxidative Stress
Increased oxidative stress is known to contribute to the pathogenesis of atherosclerosis. According to the widely accepted oxidized LDL hypothesis, atherogenicity of LDL is greatly increased by oxidative modification. Oxidatively modified LDL is taken up into monocyte/macrophages via scavenger receptors, leading foam cell formation in the subendothelial space, an early step in the atherosclerotic process. Reactive oxygen species (ROS) directly oxidize LDL, stimulate vascular smooth muscle cell proliferation and migration, and potentiate the production of proinflammatory cytokines. Reactive oxygen species activate matrix metalloproteinases, which can lead to atherosclerotic plaque instability and
rupture, as a precipitant of cardiovascular events. Reactive oxygen species also increase production of proinflammatory cytokines and acute phase proteins via activation of the transcription factor NF Kappa-B. NF-κB activation is controlled by the oxidation-reduction (redox) status of the cell, and generation of intracellular ROS may be a common step in all of the signaling pathways that lead to activation of NF-κB.
rupture, as a precipitant of cardiovascular events. Reactive oxygen species also increase production of proinflammatory cytokines and acute phase proteins via activation of the transcription factor NF Kappa-B. NF-κB activation is controlled by the oxidation-reduction (redox) status of the cell, and generation of intracellular ROS may be a common step in all of the signaling pathways that lead to activation of NF-κB.
Uremia is now well recognized as an increased oxidative stress state. Increased oxidative stress in CKD and ESRD is characterized by excess formation and retention of lipid peroxidation products, alpha, beta unsaturated reactive aldehydes, and oxidized thiols. Levels of plasma F2-isoprostanes, a lipid peroxidation endproduct, are two to four times higher in patients undergoing hemodialysis than in age- and gender-matched healthy subjects. Levels of minimally oxidatively modified LDL are also higher in patients undergoing hemodialysis than healthy subjects. Several recent studies demonstrate that plasma levels of individual plasma oxidized solutes or antioxidants can predict subsequent cardiovascular mortality in patients undergoing dialysis. A linkage between increased oxidative stress and acute-phase inflammation in severely hypoalbuminemic dialysis patients has been described. Kidney transplantation, dialysis therapy, and antioxidant therapy may each have limited but beneficial effects on oxidative stress-related metabolic abnormalities.
Similar to assessment of inflammation in kidney disease, it has now become clear that the hemodialysis procedure itself is not the predominant cause of the increased oxidative stress observed in the ESRD population. The net effect of a single hemodialysis treatment on oxidative stress parameters is variable, as many oxidized solutes are removed, while there is likely a small increase in oxidant production. Even when cellulosic dialysis membranes that vigorously activate the alternative pathway of complement are used, it is difficult to detect an increase in oxidative stress over the course of the dialysis procedure. The best available evidence suggests that overall dialysis improves redox balance but does not have a long-term effect to mitigate oxidative stress.
Although the dialysis population is better-studied, data now convincingly demonstrates that patients with less severe degrees of CKD are also subject to high levels of oxidative stress. Multiple biomarkers of oxidative stress (and inflammation) are elevated in patients with moderate CKD compared with healthy subjects. Increases in lipid peroxidation end-products, protein carbonyls, advanced oxidation protein products (AOPPs), and changes in glutathione content have all been observed. Thus, there is a surprisingly high prevalence of inflammation and oxidative stress associated with the development of mild to moderate CKD.
The pathogenesis of uremic oxidative stress is not well understood; however, leukocyte activation clearly is an important contributor. Stimulated neutrophils and monocytes generate superoxide and hydrogen peroxide (after dismutation) and release the heme enzyme myeloperoxidase (MPO) during degranulation. MPO is one of the most abundant proteins in phagocytes, constituting approximately 5% neutrophil protein and 1% monocyte protein. MPO has the unique property of converting chloride in the presence of hydrogen peroxide to hypochlorous acid. A substantial
body of evidence suggests that MPO is involved in uremic inflammation and oxidative stress. Catalytically active MPO can be released during the hemodialysis procedure, and 3-chlorotyrosine, an oxidative stress biomarker specific for MPO-catalyzed oxidation through hypochlorous acid, has been demonstrated in the plasma proteins of patients undergoing dialysis.
body of evidence suggests that MPO is involved in uremic inflammation and oxidative stress. Catalytically active MPO can be released during the hemodialysis procedure, and 3-chlorotyrosine, an oxidative stress biomarker specific for MPO-catalyzed oxidation through hypochlorous acid, has been demonstrated in the plasma proteins of patients undergoing dialysis.
MPO can also directly modulate vascular inflammatory responses by functioning as a vascular nitric oxide oxidase, thereby regulating nitric oxide availability. Because MPO is released during acute inflammation, MPO-catalyzed oxidative injury and nitric oxide regulation provide a direct mechanistic linkage between inflammation, oxidative stress, and endothelial dysfunction. Other factors that contribute to increased oxidative stress in patients with CKD include dietary deficiency of antioxidants and scavenging systems, diabetes, inflammation, and age-related changes in antioxidant defenses.
Insulin Resistance
Glucose metabolism can be impaired by defects in insulin secretion from pancreatic beta cells and/or from defects in cellular sensitivity to insulin. Each mechanism may result in hyperglycemia and diminished glucose tolerance (defined by elevated circulating glucose concentrations after an oral or intravenous glucose challenge). There are several diagnostic techniques available for the assessment of insulin resistance. Fasting glucose measurements and oral glucose tolerance tests are routinely used to clinically identify disease, e.g., for the diagnosis of type 2 diabetes. More invasive diagnostic tests can be used to define underlying pathophysiology. These tests include glucose clamp studies and the intravenous glucose tolerance test (IVGTT). During the IVGTT, glucose and insulin concentrations are measured after a standard intravenous glucose bolus. The insulin response to the glycemic load reflects insulin secretion, while modeling relates glucose disappearance to insulin concentrations to calculate insulin sensitivity. Fasting insulin concentrations or indices calculated from fasting insulin and glucose (e.g., the Homeostasis Model Assessment [HOMA]), are also frequently used to estimate insulin sensitivity under basal conditions, particularly in large epidemiologic studies. The hyperinsulinemic euglycemic clamp procedure is the criterion standard for quantifying insulin sensitivity. During administration of exogenous insulin, an infusion of glucose is titrated to a rate that maintains peripheral glucose concentration at a normal fasting level. The glucose infusion rate equals the rate of glucose utilization by the body, providing a global measure of insulin sensitivity.
Glucose metabolism is frequently impaired in CKD, predominantly because of an extraordinarily high prevalence of insulin resistance (Fig. 2-2). In ESRD, insulin resistance was demonstrated to be the most profound disturbance using euglycemic and hyperglycemic clamp techniques. Specifically, ESRD is characterized by reduced insulin-mediated glucose uptake in skeletal muscle. Some patients with ESRD are able to compensate for insulin resistance by increasing insulin secretion. However, defects in insulin secretion are also common in ESRD. A recent epidemiologic study using data from the Third National Health
 Nutritional Support in Acute Renal Failure
Nutritional Support in Acute Renal Failure
 Requirements for Protein, Calories, and Fat in the Predialysis Patient
Requirements for Protein, Calories, and Fat in the Predialysis Patient
 Nutritional Requirements in Hemodialysis Patients
Nutritional Considerations in Patients on Peritoneal Dialysis
Nutritional Requirements in Hemodialysis Patients
Nutritional Considerations in Patients on Peritoneal Dialysis
 Exercise and Physical Function in Kidney Disease
Nutritional Interventions in Chronic Kidney Disease
Exercise and Physical Function in Kidney Disease
Nutritional Interventions in Chronic Kidney Disease




Related posts:
Nutritional Support in Acute Renal Failure
Requirements for Protein, Calories, and Fat in the Predialysis Patient
Nutritional Requirements in Hemodialysis Patients
Nutritional Considerations in Patients on Peritoneal Dialysis
Exercise and Physical Function in Kidney Disease
Nutritional Interventions in Chronic Kidney Disease
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
