A recent definition states that idiosyncratic reactions are toxic responses determined by individual susceptibility to (host) factors that increase the penetrance and expressivity of the intrinsic toxicity of a drug (or a drug metabolite) [26]. Indeed, development of DILI is a complex, multi-step process in which the toxic potential of the drug, genetic and acquired factors, as well as individual deficiencies in the adaptive processes, that limit the extent of the injury, determine the susceptibility to the rare occurrence of idiosyncratic hepatotoxicity (see Figure 47.1) [27]. Thus, most patients tolerate the drug without adverse liver effects or there is a background of mild, and often transient, asymptomatic liver injury (i.e. statins, isoniazid), indicating an adaptation to the drug and further tolerance. Therefore, the combination of susceptibility factors coupled with drugs that (due to variation in handling between different phases of drug metabolism, detoxification, and transport) reach a threshold for exposure to drug or toxic metabolites, enhances the risk of idiosyncratic hepatotoxicity.
Several mechanisms and pathways may be involved in DILI. Most frequently, damage is initiated through the formation of reactive drug metabolites that can be directly toxic or induce immune reactions [28,29]. In other instances drugs can impair mitochondrial function, which may decrease fat oxidation (causing fat accumulation) and/or energy production leading to cell death [30, 31]. Also parent drugs or reactive drug metabolites, either through direct toxicity or immune reactions, may open the mitochondrial permeability transition pore, which subsequently results in necrosis or apoptosis [32]. A unifying hypothesis that involves underlying genetic or acquired mitochondrial abnormalities as a major determinant of susceptibility for a number of drugs that target mitochondria and cause DILI has been proposed [26]. The mitochondrial hypothesis requires gradually accumulating and initially silent mitochondrial injury in heteroplasmic cells, which then reaches a critical threshold and abruptly triggers liver injury. This is consistent with the fact that, typically, idiosyncratic DILI is delayed (by weeks or months). New animal models (e.g. the Sod2+/– mouse) provide supporting evidence for this concept [26]. However, genetic analyses of DILI patient samples are ultimately needed to provide evidence for this proof-of-concept [26].
In drug-induced immune-mediated hepatic injury, the drug reactive metabolites trigger an adaptive immune response directed against the drug-modified liver components, resulting in acute or chronic liver damage. Advances in the understanding of the pathogenesis of hepatotoxicity are hampered by the fact that no satisfactory animal models for idiosyncratic DILI have been developed. Recent evidence in experimental acetaminophen hepatotoxicity clearly indicates that the balance between the pro-and anti-inflammatory mediators determines the susceptibility and severity of liver injury [33]. Conceivably, these insights could be extrapolated to idiosyncratic DILI in humans, particularly the role of the innate immune system and cell-death pathways.
Risk factors
Most of the risk factors that have been identified are the ones which influence drug disposition and metabolism. Incomplete information in case reports has precluded analyses of drug-related, environmental risk factors and clinical presentation profiles, which, however, can be obtained in large cohorts of patients with DILI [21,22,34–37]. Known risk factors are, nevertheless, poorly predictive of hepatotoxicity, and in many instances of DILI no risk factor can be identified [38].
Environmental factors
There are several environmental factors that appear to operate in determining individual susceptibility.
Age
Elderly patients are more vulnerable to DILI. In particular, increased age is a susceptibility factor for developing hepatotoxicity to antituberculous drugs [39]. Older age has also been implicated in amoxicillin-clavulanate induced cholestasis [40, 41]. In contrast, valproic acid and erythro-mycin hepatotoxicity are more common in childhood [42]. Older age, rather than being a predisposing factor to DILI, seems to have an impact on the phenotypic expression of toxic liver damage, strongly favoring the appearance of a cholestatic pattern of injury [21, 43].
Gender
Women are more susceptible than men to most forms of DILI [18, 44–46]. For instance, autoimmune hepatitis triggered by drugs is seen almost exclusively in women [47], and diclofenac hepatotoxicity has been reported more frequently in women with osteoarthritis [42].
However, new epidemiological data have challenged this traditional belief, showing a similar sex distribution in DILI cases [21, 48], with a higher prevalence of female gender only at younger ages [43]. Yet, female gender is considered to be a risk factor for developing fulminant liver failure [21], and female sex accounted for 76% of patients presenting with drug-induced acute liver failure in the USA who were transplanted [49]. Similarly to age, gender may influence the clinical presentation of DILI as women are over represented in the hepatocellular type of injury [43].
Chronic alcohol consumption
A history of alcohol intake was considered to be a general risk factor for idiosyncratic DILI, and as such scores 1 + point in the standard clinical scale for assignation of causality developed by experts, the CIOMS (Council for International Organizations of Medical Sciences) or RUCAM (Roussel Uclaf Causality Assessment Method) scale [50]. However, for most of the drugs capable of inducing hepatotoxicity there is no evidence for a role of alcohol in potentiating toxicity. In practical terms, chronic alcohol intake increases the risk of developing liver fibrosis during methotrexate therapy, enhances acetaminophen hepatotoxicity by inducing cytochrome P-450 (CYP) 2E1 (with generation of higher levels of the reactive metabolite and depletion of glutathione stores), as well as susceptibility to liver damage from isoniazid, halothane and cocaine [42].
Concomitant drugs
These are capable of modulating the hepatotoxic potential of other drugs through CYP or hepatic transport system induction, inhibition or substrate competition. In addition, concomitant medication with hepatotoxic potential may further increase the risk [51]. Concurrent use of anticonvul-sants which induce drug metabolism greatly increases the risk of hepatotoxicity due to valproate [42]. The concurrent use of isoniazid and rifampicin, a potent microsomal inducer, enhances the risk of isoniazid hepatotoxicity [52].
Underlying disease states
Controversy exists as to whether pre-existing liver disease is a susceptible factor for the development of hepatotoxicity. The presence of chronic hepatitis B, hepatitis C infection or co-infection with human immunodeficiency virus (HIV) increases the risk of isoniazid hepatotoxicity or elevated transaminases on HAART (highly active antiretrovi-ral therapy) [53]. The increased susceptibility exhibited by HIV patients suggests a role for cytokine imbalance [54]. Rifampicin is more hepatotoxic when used for control of itching in patients with primary biliary cirrhosis [55].
Obesity, diabetes mellitus type 2 and insulin resistance are known risk factors for steato-hepatitis, and have also been shown, along with psoriasis, to increase the risk of developing liver fibrosis during methotrexate therapy [56].
Genetic risk factors
Idiosyncratic hepatotoxicity is not predictable based either on the pharmacology of the drug or the dose administered, even when confounding variables such as environmental factors are taken into account. On the other hand, recurrence of liver injury with re-exposure to the drug often occurs under different clinical circumstances, indicating that susceptibility to hepatotoxicity could depend principally on host genetic factors [57]. Association studies have sought for single nucleotide polymorphisms (SNPs) in candidate genes, which are those presumably involved in drug bioactivation/detoxification processes as well in the immune response (see Table 47.1).
The reactive metabolite hypothesis states that most DILI instances would result from the production of reactive metabolites [20, 28, 29, 58, 59], which accumulate within the hepatocyte to levels that exceed a critical threshold. Once this threshold is crossed the damage is initiated. In liver, CYPs are more abundant in the centrilobular (zone 3) than periportal regions. Therefore, centrilobular necrosis, which represents one typical histological feature of serious acute drug-induced liver injury, may indeed indicate that drug metabolizing enzymes have a major role in pathogenesis. Only anecdotal reports suggest a role of CYP 450 polymorphisms in hepatotoxicity. For example, CYP2D6 deficiency is associated with perhexiline hepatotoxicity [60], and may occur in up to 10% of Europeans [61]. CYP 2C19 deficiency was suggested to be associated with Atrium (a phenobarbitone containing combination preparation) liver injury [62]. However, 60 patients with DILI caused by drugs known to be substrates of CYP2C9 and CYP2C19, showed a similar prevalence of allelic variants to those in other European populations, and there were no patients exhibiting very low enzyme activity with CYP2C9 *3/*3 and CYP2C19 *3/*3 genotype [60]. These data suggest that CYP2C9 and CYP2C19 genetic polymorphisms might not be a predictable potential risk factor for DILI. Indeed, variant genotypes of CYP, associated with lower enzyme activity could not account for higher susceptibility to DILI, as an intact or even enhanced enzyme activity (determined by environmental rather than genetic factors) seems to be required for the formation of reactive metabolites.
An alternative possibility is that reactive metabolites may not undergo detoxification, either because they are poor substrates, or because of failure of detoxification enzyme function (due to genetic polymorphisms). Phase II enzymes are known to be polymorphically expressed. The major Phase II genes implicated in hepatotoxicity are N-acetyltransferase 2 (NAT2) and the glutathione S-transferases M and T (GSMT and GSTT).
In Chinese patients on antituberculosis therapy, carriers of NAT2 genotypes which are associated with slow acetyla-tion, had four-fold risk of developing isoniazid-induced hepatotoxicity [63]. Regular monitoring of serum ami-notransferase was thus suggested in slow NAT2 acetylators receiving isoniazid treatment [64]. The same authors showed that subjects with CYP2E1 c1/c1, were 2.5 times more likely to develop hepatotoxicity when compared with the other genotypes. The odds ratio for hepatotoxicity increased to 7.4 when CYP2E1 c1/c1 was combined with slow acetylator status [65].
Oxidative stress induced by the reactive metabolites or reactive oxygen species generated by drugs, may be the final common pathway underlying DILI. Individual vulnerability to a drug might then not be closely related to the rate of parent drug metabolism, but related to deficiencies in the detoxification process, or in drug transporters which ultimately determine the level of exposure to the reactive metabolite. In this regard, glutathione S-transferase (GST), a major phase II family of conjugation enzymes, prevents the binding of reactive metabolites to cellular proteins and modulates the by-products of oxidative stress, catalyzing the conjugation of electrophilic moieties to glutathione [66].
The importance of GST as a general detoxification mechanism in DILI is underlined by the fact that it acts for a variety of metabolites derived from aflatoxin, acetaminophen, benzo(a)pyrene, bromobenzene, or felbamate, and other substances [67–72]. In humans, the activity of the cytosolic GSTs M1 (μ) and T1 (θ) are polymorphically expressed due to complete GSTM1 and GSTT1 gene deletions, that occur in homozygosity (null genotypes) in 50%, and 10–25% of Caucasian subjects respectively [73]. A significant association with GSTM1-GSTT1 null genotype was found in Japanese patients with troglitazone induced DILI [74], and in a cohort of French [75], but not British patients [76], with tacrine-induced DILI. An association with GSTM1 (but not GSTT1) null genotype, was also identified in Indian patients with antituberculosis drug-induced hepatotoxicity (52% vs 24% in controls, p < 0.05) [77], and in Chinese patients with antituberculosis drug-induced hepatotoxicity, (2.23 fold-increased risk) [78]. Taken together, and consistent with studies in animal models, these findings point to a role for these enzyme activities as a general protective mechanism against hepatotoxicity. This hypothesis was tested in a study involving 154 patients with a diagnosis of DILI due to an array of medications. Carriers of double GSTT1-M1 null genotypes were 2.70 times more likely to develop hepatotoxicity, when compared with non-carriers. The odds ratio for DILI patients receiving antibacterials, and NSAIDs were 3.52 and 5.61 respectively. Patients with amoxicillin-clavulanate hepatotoxicity had a 2.81-fold increased risk. There was a statistically significant predominance of women in the combined null genotype (p < 0.001) [79].
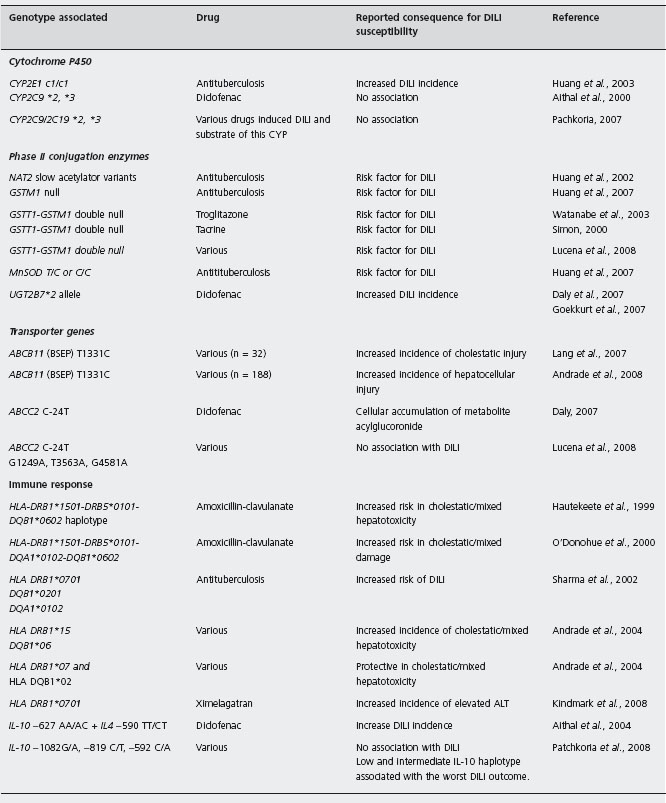
The Manganese Superoxide Dismutase (MnSOD) is also a critical determinant of cellular defence against toxic insult to the liver. A study including 115 subjects with DILI (63 with antituberculosis hepatotoxicity) showed that those with a variant C allele (genotype T/C or C/C) of MnSOD had an increased risk of hepatotoxicity compared to those with MnSOD T/T genotype, considering all drugs together (adjusted OR: 2.44), but also in the sub-category of anti-tuberculosis drugs (adjusted OR: 2.47) [78].
The functions of ATP-Binding-Cassette (ABC) transporters, which determine the excretion or accumulation of certain reactive metabolites, make them obvious candidate genes in the identification of susceptible individuals to develop DILI. In a study a single nucleotide polymorphism in exon 13 of ABCB11,1331T > C (V444A), which is associated with decreased hepatic BSEP expression, was significantly more frequent in drug-induced cholestasis than in hepatocellular injury, or in the control population [80]. In contrast, carriers of the ABCB11,1331T > C polymorphism were found to have a two-fold increased risk of developing toxic hepatocellular damage in a cohort of 188 Spanish DILI patients [81].
With the availability of increasing information on new polymorphisms and their functional significance, it is currently feasible in some cases to perform genotyping for different polymorphisms concomitantly. A study involving 24 patients with diclofenac-induced liver injury showed that simultaneous possession of variant of UDP-glucuronosyl-transferase (that mediates the glucuronocon-jugation of diclofenac to a paradoxically reactive metabolite acyl-glucuronide) the UGT2B7*2 allele (associated with increased activity), and the ABCC2 (that transports the glu-curonide to the biliary canaliculus), C-24T variant (associated with reduced activity) was more common in DILI patients compared with controls. Haplotype distributions for CYP2C8 (which plays a role in the metabolism of diclofenac to 5-hydroxydiclofenac) were also significantly different in patients with hepatotoxicity [36]. Interestingly, in this study, 21% of the community control population was positive for both the “at-risk” UGT2B7 and ABCC2 genotypes [36]. It is clear that the incidence of serious hepatotoxicity is much lower than these figures. Indeed, genetic polymorphisms of drug metabolism, although increasing the risk of DILI, do not explain why all patients with a given polymorphism do not develop DILI when treated with the relevant drug.
Immunological factors
Once hepatic cells become stressed by the toxic insult, the response of the innate and acquired immune system (downstream events), appear to determine whether liver injury subsides or progresses to clinical disease (see Figure 47.1) [27].
Variations in the production of immunoregulatory cytokines (genetic polymorphisms) such as IL-10 and IL-4 have also been reported to contribute to the susceptibility to diclofenac hepatotoxicity [82]. The observed polymorphisms, resulting in low IL-10 and high IL-4 gene transcription, could favour a Th-2 mediated antibody response to neoantigenic stimulation associated with disease susceptibility.
As the increased susceptibility related to these downstream events may be less drug-specific than those involved in the accumulation of reactive metabolites, it might be possible to group cases of DILI associated with a heterogeneous group of drugs. In a cohort of 146 patients with DILI the analysis of three polymorphisms: –1082G/A, –819C/T, and –592C/A in the IL-10 gene promoter, did not reveal any association with the predisposition to develop hepatotoxicity. However, the low IL-10 producing haplotype was more prevalent in DILI patients coupled with the absence of peripheral blood eosinophilia [83]. All cases with serious DILI outcome carried low or intermediate IL-10 producing haplotypes and had normal or low eosinophil counts. [83] Indeed, low IL-10 producing haplotype, leading to a reduced or absent rise in peripheral eosinophil count, could be a marker of an unfavorable outcome of DILI, supporting the concept of accelerated Th1 T-cytotoxic response.
HLA genotyping
Major histocompatibility complex molecules (i.e. class I and II HLA molecules) participate in antigen presentation to immunocompetent cells and in the regulation of the immune response. Significant association has been reported between DRB1*1501-DRB5*0101-DQB1*0602 haplotype and cholestatic hepatitis related to amoxicillin-clavulanate [84]. Also a genome-wide association study using 866,399 markers in 51 cases of flucloxacillin DILI and 282 controls matched for sex and ancestry showed an association peak in the major histocompatibility complex (MHC) region with the strongest association (P = 8.7 x 10(–33)) seen for rs2395029[G], and other SNPs all of which are a part of an extended MHC 57.1 haplotype present in <4% of Europeans, which is associated to hypersensitivity to abacavir [85]. However, even in cases of DILI with no evidence of allergy, such as those induced by ximelagatran (hepatocellular type, delayed in latency, no eosinophilia), using a wide genome screen approach, a strong genetic association between elevated alaninoaminotransferases (ALT) and the HLA class-II alleles DRB1*07 and DQA1*02 was also recently shown, suggesting a possible immune pathogen-esis. Consistent with this hypothesis, immunological studies suggest that ximelagatran may have the ability to act as a contact sensitizer, and hence be able to stimulate an adaptive immune response [86].
A large cohort of cases with drug-induced liver disease due to variety of agents with a wide range of manifestations may be suitable in the evaluation of the effect of genetic factors on the phenotype expression of hepatotoxic-ity [87]. The HLA DRB1*15 and DQB1*06 alleles of the class II HLA system participate in increased susceptibility to the development of a cholestatic/mixed pattern in drug-induced liver injury, thus providing indirect evidence about the immunoallergic nature of this specific pattern of damage and explaining, at least in part, why a given drug may cause different patterns of liver damage [87].
Diagnostic approach and tools for causality assessment
The phenotypic expressions of DILI may present in several ways (clinical and pathological) that resemble known forms of acute and chronic liver disease; the severity ranges from sub-clinical elevations in liver enzyme concentrations to acute liver failure. Mainly, drugs tend to induce acute hepatitis, cholestasis or a mixed condition (see Table 47.2) [47].
Liver histology (although not very specific and, at best, “compatible with”) is still the ideal tool for defining the pattern of liver damage. However, since a liver biopsy specimen is often not available, the pattern of drug-related liver injury is, from a practical standpoint, classified according to laboratory data [88, 89]. The classification scheme was proposed by CIOMS [88] and recently updated by the Food and Drug Administration Drug Hepatotoxicity Committee (see Table 47.3) [89].
The acute hepatocellular (cytotoxic, cytolytic) type of liver injury is the most common expression of hepatotoxicity and it is observed with many drugs (see Table 47.4) [21, 22]. Patients with acute hepatocellular injury related to drugs are at risk of acute liver failure. The presence of combined increases in ALT and bilirubin levels in DILI reflects a substantial loss in hepatocellular function and potential for liver failure [90]. B4 These parameters are considered a specific indicator of severe hepatotoxic potential of a drug. In patients with acute drug-induced hepatitis the presence of jaundice is the most significant predictor of mortality /liver transplantation. The observation by Hyman Zimmerman, known as “Hy’s rule” [47], predicts a mean mortality (or its surrogate marker, liver transplantation) of 10% (range 5–50%) for jaundiced patients with acute toxic hepatocellular damage. Hy’s rule has been validated in large case series from Spain and Sweden in which analysis of pooled data showed that other variables such older age, female gender and AST levels were independently associated with a poor outcome [21, 38]. B4 Further analysis of the Swedish database, has found that there was also a significant linear relationship between daily dose and the outcome of death/liver transplantation (2%, 9.4% and 13.2% in –10, 11-49, and >50mg/day groups respectively, p = 0.03) [24]. B4 On the contrary, it has recently been suggested that eosinophilia accompanying DILI may be associated with a better short-term prognosis [91, 92]. Actually, none of the patients included in the Spanish Registry with idiosyncratic DILI who died, evolved to liver failure, or required a liver transplantation had eosinophilia, whereas this feature was found in the 23% of the patients with milder outcomes [21]. B4 Of note is that the height of transaminases in acute hepatocellular DILI lacks prognosis significance. Indeed, a decrease in liver enzymes after drug withdrawal in patients with severe DILI may reflect rather than a clinical improvement, a limited hepatic reserve associated with impending liver failure [90]. Moreover, if acute injury is superimposed on underlying liver disease, monitoring liver function tests may be difficult to interpret. For example, if advanced cirrhosis is present, the severity of liver injury may be underestimated by the height of the serum ALT measurements [93].
Table 47.2 Key clinical aspects in drug-induced liver disease.
| DILI clinical issue | Key point | To be remembered |
| Presentation | • DILI may resemble any acute and chronic liver disease. | • Hypersensitivity features occur in less than a quarter of patients. |








