Disorders of the Adrenal Glands: Introduction
Disorders of the adrenal glands result in classic endocrine syndromes such as Cushing’s syndrome, hyperaldosteronism, and catechol excess from pheochromocytoma (Figure 31–1). The diagnosis of these disorders requires careful endocrine evaluation and imaging with computed tomography (CT) or magnetic resonance imaging (MRI).
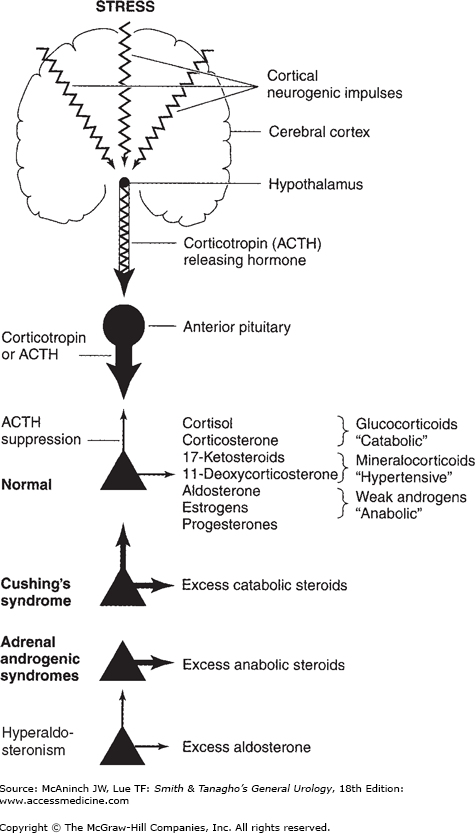
Diseases of the Adrenal Cortex
Cushing’s syndrome is the clinical disorder caused by overproduction of cortisol. Most cases (80%) are due to bilateral adrenocortical hyperplasia stimulated by overproduction of pituitary adrenocorticotropic hormone (ACTH, corticotropin), known as Cushing’s disease. About 10% of cases are due to the ectopic production of ACTH from nonpituitary tumors. Ectopic ACTH production occurs most frequently in small-cell lung carcinoma; other tumors producing ACTH include carcinoids (lung, thymic, gastrointestinal tract), islet cell tumors of the pancreas, medullary thyroid carcinoma, pheochromocytoma, and small-cell carcinoma of the prostate. Adrenal adenoma is the cause in 5% of cases and carcinoma in 5%. In children, adrenocortical carcinoma is the most common cause of Cushing’s syndrome.
Overproduction of cortisol by adrenocortical tissue leads to a catabolic state. This causes liberation from muscle tissue of amino acids, which are transformed into glucose and glycogen in the liver by gluconeogenesis. The resulting weakened protein structures (muscle and elastic tissue) cause a protuberant abdomen and poor wound healing, generalized muscle weakness, and marked osteoporosis, which is made worse by excessive loss of calcium in the urine.
In addition, glucose is transformed largely into fat and appears in characteristic sites such as the abdomen, supraclavicular fat pads, and cheeks. There is a tendency to diabetes, with an elevated fasting plasma glucose level in 20% of cases and abnormal glucose tolerance in 80% of patients.
The cortisol excess also suppresses the immune mechanisms, making patients susceptible to infection. Inhibition of fibroblast function by excess cortisol further interferes with wound healing.
Hypertension is present in 90% of cases. Although the aldosterone level is not usually elevated, cortisol itself exerts a hypertensive effect when present in excessive amounts, as does 11-deoxycorticosterone. The hypertension may be accompanied by manifestation of mineralocorticoid excess (hypokalemia and alkalosis), especially in patients with ectopic ACTH syndrome or adrenocortical carcinoma.
The cells in adrenal hyperplasia resemble those of the zona fasciculata of the normal adrenal cortex. Frank adenocarcinoma reveals pleomorphism and invasion of the capsule, the vascular system, or both (Figure 31–2). Local invasion may occur, and metastases are common to the liver, lungs, bone, or brain. Histologic differentiation between adenoma and adenocarcinoma is frequently difficult.
Figure 31–2.
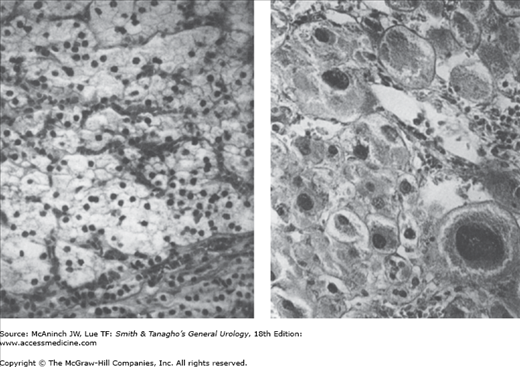
Left: Histologic appearance of a typical benign adenoma of the adrenal cortex made up of a large number of identical cells from the zona fasciculata removed from a 39-year-old woman with Cushing’s syndrome. Right: Section of an adenocarcinoma removed from a 36-year-old woman with metastatic adenocarcinoma showing significant pleomorphism of the cells. Invasion of a large vein is not shown in this micrograph. Note that benign adenomas will occasionally have this appearance but without invasion of the bloodstream. (Reproduced, with permission, from Forsham PH: The adrenal cortex. In: Williams RH [ed.] Textbook of Endocrinology. 4th ed. Saunders, 1968.)
In the presence of adenoma or malignant tumor, atrophy of the cortices of both adrenals occurs because the main secretory product of the tumor is cortisol, which inhibits the pituitary secretion of ACTH. Thus, although the tumor continues to grow, the contralateral adrenal cortex undergoes atrophy.
The presence of at least three of the following strongly suggests Cushing’s syndrome:
Obesity (with sparing of the extremities), moon face, and fat pads of the supraclavicular and dorsocervical areas (buffalo hump).
Striae (red and depressed) over the abdomen and thighs.
Hypertension (almost always present).
Proximal myopathy with marked weakness, especially in the quadriceps femoris, making unaided rising from a chair difficult.
Emotional lability, irritability, difficulty in sleeping, and sometimes psychotic personality.
Osteoporosis (common), with back pain from compression fractures of the lumbar vertebrae as well as rib fractures.
In 80% of cases, postprandial hyperglycemia is present, and in 20%, there is an elevated fasting plasma glucose level.
To a variable extent, there are features of adrenal androgen excess in women with Cushing’s syndrome; these are absent in the case of adenoma, most severe with carcinoma, and present to an intermediate degree with Cushing’s disease. They consist of recession of the hairline, hirsutism, small breasts, and generalized muscular overdevelopment, with deepening of the voice.
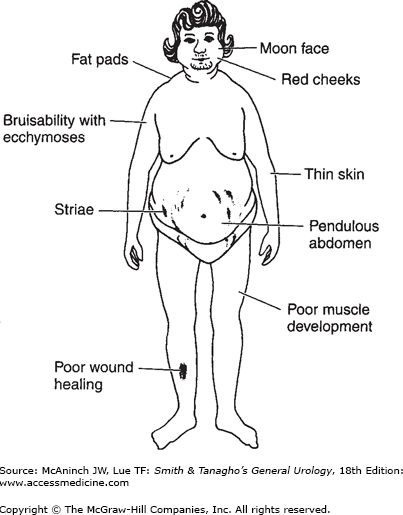
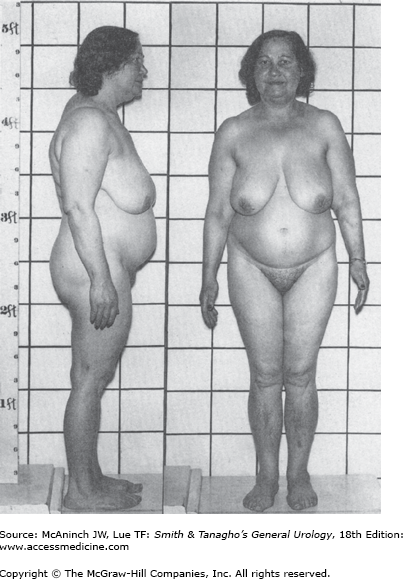
It is not possible to differentiate the cause from the clinical presentation alone.
The leukocyte count may be elevated to the range of 12,000–20,000/μL, usually with fewer than 20% lymphocytes. Polycythemia is present in over half the cases, with the hemoglobin ranging from 14 to 16 g/dL. Anemia, however, may occur in patients with malignant tumors ectopically secreting ACTH.
Blood chemical analyses may show an increase in serum Na+ and CO2 levels and a decrease in serum K+ levels. Hyperglycemia may occur.
The following tests are performed to determine whether the patient has Cushing’s syndrome.
24-Hour urinary cortisol level—Urine cortisol is measured in a 24-hour urine collection (normal range, 10–50 μg/24 h). A urine cortisol value more than twofold elevated is typical of Cushing’s syndrome. False-positive elevations can occur in acute illness, depression, and alcoholism. However, obesity does not raise the level of urinary-free cortisol above normal.
Suppression of ACTH and plasma cortisol by dexamethasone—Dexamethasone in low doses is used to assess the feedback suppression of ACTH and cortisol production by glucocorticoids. If dexamethasone is given at 11 pm, ACTH is suppressed in normal persons but not in those with Cushing’s syndrome. Dexamethasone is useful because it has 30 times the potency of cortisol as an ACTH suppressant and it is not measured in current plasma or urine cortisol methods.
The procedure is to give 1 mg of dexamethasone by mouth at 11 pm and to draw blood at 8:00 to 9:00 am for measurement of plasma cortisol. If the level is <5 μg/dL (normal is 5–20 mg/dL), Cushing’s syndrome can be ruled out. If the value is >10 μg/dL, Cushing’s syndrome is present. A level in the range of 5–10 μg/dL is equivocal, and the test should be repeated or the urine cortisol may be measured.
Women taking birth-control pills have high plasma cortisol levels because, as in pregnancy, the estrogen stimulates production of cortisol-binding globulin. The pills must be withheld for at least 3 weeks before the dexamethasone suppression test. Other conditions causing false-positive responses are acute illness, depression, and alcoholism. Also, about 15% of obese patients do not suppress cortisol with this test.
The various causes of Cushing’s syndrome can be determined with great accuracy (95% of cases).
Plasma ACTH level—If the diagnosis of Cushing’s syndrome has been established, this test will differentiate ACTH-dependent causes (Cushing’s disease and the ectopic ACTH syndrome) from adrenal tumors, which are ACTH independent. The normal range is 10–50 pg/mL. Patients with Cushing’s disease have ACTH levels that range from 10 to 200 pg/mL; in the ectopic ACTH syndrome, levels are usually >200 pg/mL; and patients with adrenal tumors have suppressed ACTH levels (<5 pg/mL).
Plasma androgen levels—In patients with adrenal adenomas, androgen levels are normal or low, and in adrenocortical carcinoma, these levels are often markedly elevated.
When tests suggest Cushing’s disease or the ectopic ACTH syndrome and an elevated plasma level of ACTH is present, the source of ACTH must be identified. Because the great majority of these patients have Cushing’s disease and because most of the patients with ectopic ACTH secretion have an obvious malignancy, the first step is to perform pituitary MRI. These are positive in 50–60% of patients with Cushing’s disease; in the remainder, the diagnosis should be established by the sampling of ACTH levels in the venous drainage of the anterior pituitary, that is, the cavernous sinuses and inferior petrosal sinuses. If the MRI and venous sampling do not reveal a pituitary source of ACTH, CT scans of the chest and abdomen are used to localize an ectopic tumor.
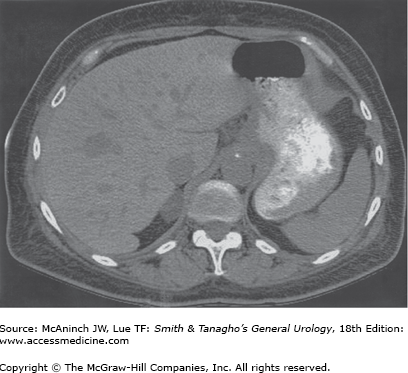
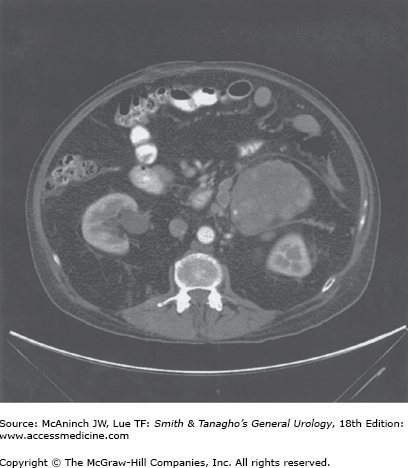
Patients with Cushing’s syndrome with suspected adrenal tumors and suppressed ACTH levels should undergo a CT scan of the abdomen with 3-mm sections through the adrenals. Adrenal tumors causing Cushing’s syndrome are usually >3 cm in diameter (Figure 31–5) and are therefore easily visualized. Adenomas are usually 3–6 cm in diameter; carcinomas are usually >5 cm in diameter (Figure 31–6) and are frequently locally invasive or metastatic to the liver and lungs at the time of diagnosis. In patients with adrenal tumors, the contralateral adrenal is suppressed and therefore appears atrophic or normal on CT scan. The finding of bilateral adrenal enlargement is typical of Cushing’s disease or the ectopic ACTH syndrome. Ultrasound or MRI may also be used for adrenal localization, although these techniques do not appear to offer significant advantage over CT.
A pituitary microadenoma, which is the most common cause of bilateral adrenocortical hyperplasia, must be located and removed surgically. Transsphenoidal resection performed by an experienced neurosurgeon is the method of choice. Success is reported in >80% of cases, and in most instances, the endocrine functions of the pituitary gland are preserved.
The treatment of these patients is difficult because most have an advanced malignancy and severe hypercortisolism. Removal of the primary tumor is clearly the therapy of choice; however, curative resection is limited to the few patients with benign tumors such as bronchial carcinoids. Patients with residual or metastatic tumors should be managed first with adrenal inhibitors, and if that is not successful, bilateral adrenalectomy should be considered.
Total bilateral adrenalectomy is indicated in patients with Cushing’s disease in whom the pituitary tumor is not resectable and in whom medical therapy fails to control the cortisol excess. A laparoscopic approach to adrenalectomy is preferred, since it significantly decreases morbidity and length of hospital stay compared with open adrenalectomy. Bilateral adrenalectomy is also indicated in patients with ectopic ACTH syndrome who have life-threatening hypercortisolism that cannot be controlled by inhibitors of adrenal secretion.
Because removal of the source of excessive cortisol will inevitably lead to temporary or permanent adrenal insufficiency, it is of the utmost importance to administer cortisol preoperatively and to continue substitution therapy after surgery to control Addison’s disease. In the postoperative period, the dose is tapered downward until oral medication provides sufficient control.
The patient feels moderately well following removal of the source of excess ACTH or adrenalectomy or while receiving a high dose of hydrocortisone in excess of the usual daily output of approximately 20 mg. It is important to reduce the steroid substitution gradually over a period of several days. On the day of operation, 200 mg of cortisol is given; the dosage is then reduced gradually on successive days (150, 100, 80, 60, and 40 mg) until a maintenance dosage of 20–30 mg cortisol combined with 0.1 mg fludrocortisone is reached.
Virtually all adrenal adenomas and smaller adrenal carcinomas are now removed laparoscopically, again allowing decreased hospital stay and more rapid recovery from surgery. Most patients now have a 1-day hospitalization after laparoscopic adrenalectomy if their metabolic management allows. There is some emerging evidence that select small adrenal tumors can be percutaneously ablated with radiofrequency ablation (RFA). The long-term efficacy of this approach is not known. Adrenal carcinomas that are large, locally invasive or involve the inferior vena cava are best approached by an open operation.
Preoperative preparation is the same as that for bilateral hyperplasia, since, in this case, the remaining adrenal gland will be atrophic and thus the patient will be hypoadrenal.
Cortisol is administered perioperatively in the doses described earlier and then tapered to a replacement dose of 20–30 mg/day. Hydrocortisone is given orally in a dosage of 10 mg three times daily initially and reduced within 2–3 weeks to 10 mg daily given at 7 or 8 am. Substitution therapy may be necessary for 6 months to 2 years depending on the rate of recovery of the residual gland. Mineralocorticoid therapy is rarely necessary, since the atrophic adrenal usually produces sufficient aldosterone. Patients with adrenocortical carcinoma are usually not cured by surgery and require additional therapy.
There is no effective method of inhibiting ACTH secretion; however, adrenal hypersecretion can be controlled in many patients by inhibitors of adrenal cortisol secretion. Medical therapy is indicated in patients who either cannot undergo surgery (eg, because of debility, recent myocardial infarction) or in those who have had unsuccessful resection of their pituitary, ectopic, or adrenal tumor.
Ketoconazole is the current drug of choice; it blocks cortisol secretion by inhibiting P450c11 and P450scc. The total dose required is 800–1600 mg/day given in two divided doses. Side effects are adrenal insufficiency, abnormal liver function tests, and hepatotoxicity in a few patients.
Metyrapone may be used alone or may be added if ketoconazole alone is unsuccessful in normalizing cortisol levels. The usual dosage is 1–4 g daily given in four divided doses.
Aminoglutethimide and trilostane also inhibit adrenal secretion, but they are uncommonly used at present.
Mitotane (o,p9´-DDD, Lysodren) is both an inhibitor of adrenal secretion and a cytotoxic agent that damages adrenocortical cells. It is used almost exclusively in patients with residual adrenocortical carcinoma, in whom it helps to reduce cortisol hypersecretion. The usual dosage is 6–12 g daily in three to four divided doses. About 70% of patients achieve a reduction in steroid secretion and 35% achieve a reduction in tumor size; however, there is no convincing evidence that the drug prolongs survival. Side effects occur in 80% of patients and include nausea, vomiting, diarrhea, depression, and somnolence.
Treatment of hypercortisolism usually leads to disappearance of symptoms and many signs within days to weeks, but osteoporosis usually persists in adults, whereas hypertension and diabetes often improve. Cushing’s disease treated by pituitary adenomectomy has an excellent early prognosis, and long-term follow-up shows a recurrence rate of about 10%. Patients with the ectopic ACTH syndrome and malignant tumors in general have a poor prognosis. Patients with benign lesions may be cured by resection of the tumor. Removal of an adrenal adenoma offers an excellent prognosis, and these patients are cured by unilateral adrenalectomy.
The outlook for patients with adrenocortical carcinoma is poor. The antineoplastic drug mitotane reduces the symptoms and signs of Cushing’s syndrome but does little to prolong survival. Radiotherapy and chemotherapy are not successful in these patients.
Adrenal androgenic syndromes are more common in females. Congenital bilateral adrenal hyperplasia and tumors, both benign and malignant, may be observed. In contrast to Cushing’s syndrome, which is protein catabolic, the androgenic syndromes are anabolic. In untreated cases, there is a marked recession of the hairline, increased beard growth, and excessive growth of pubic and sexual hair, in general, in both sexes. In males, there is enlargement of the penis, usually with atrophic testes; in females, enlargement of the clitoris occurs, with atrophy of the breasts and amenorrhea. Muscle mass increases and fat content decreases, leading to a powerful but trim figure. The voice becomes deeper, particularly in females; this condition is irreversible, because it is due to enlargement of the larynx. In both sexes, there may be increased physical sexual aggressiveness and libido.
A congenital defect in certain adrenal enzymes results in the production of abnormal steroids, causing a disorder of sexual differentiation (DSD) in females and macrogenitosomia in males. The enzyme defect is associated with excess androgen production in utero. In females, the Müllerian duct structures (eg, ovaries, uterus, and vagina) develop normally, but the excess androgen exerts a masculinizing effect on the urogenital sinus and genital tubercle, so that the vagina is connected to the urethra, which, in turn, opens at the base of the enlarged clitoris. The labia are often hypertrophied. Externally, the appearance is that of severe hypospadias with cryptorchidism.
The adrenal cortex secretes mostly anabolic and androgenic steroids, leading to various degrees of cortisol deficiency depending on the nature of the enzyme block. This increases the secretion of ACTH, which causes hyperplasia of both adrenal cortices. The cortices continue to secrete large amounts of inappropriate anabolic, androgenic, or hypertensive steroids. Absence or reduction of the usual tissue concentration of various enzymes accounts for blocks in the adrenocortical synthetic pathways. 21-Hydroxylase deficiency, which is the most common cause of congenital adrenal hyperplasia, does not allow for the transformation of 17α-hydroxyprogesterone to cortisol. This common deficiency occurs in two forms: the salt-losing variety, with low-to-absent aldosterone, and the more frequent non–salt-losing type. Infants present with adrenal insufficiency and DSD; older children develop pseudoprecocious puberty and accelerated growth and skeletal maturation. Other enzymatic defects cause specific defects in sexual differentiation, adrenal insufficiency, hypertension, or salt wasting.
In newborn girls, the appearance of the external genitalia resembles severe hypospadias with cryptorchidism. Infant boys may appear normal at birth. The earlier in intrauterine life the fetus has been exposed to excess androgen, the more marked the anomalies.
In untreated cases, hirsutism, excess muscle mass, and, eventually, amenorrhea are the rule. Breast development is poor. In males, growth of the phallus is excessive. The testes are often atrophic because of inhibition of gonadotropin secretion by the elevated androgens. On rare occasions, hyperplastic adrenocortical rests in the testes make them large and firm. In most instances, there is azoospermia after puberty.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


