CHAPTER 111 Crohn’s Disease
Idiopathic inflammatory bowel disease (IBD) comprises conditions characterized by chronic or relapsing immune activation and inflammation within the gastrointestinal tract. Crohn’s disease and ulcerative colitis (UC) are the two major forms of idiopathic IBD; less common, but increasingly recognized, are the microscopic colitides, primarily collagenous colitis and lymphocytic colitis (see Chapter 124). Other chronic inflammatory conditions of the intestine share some features of presentation and pathogenesis with idiopathic IBD, but they have identifiable etiologies. These disorders include diversion colitis, bypass enteropathy, radiation colitis, and drug-induced colitides. The two major forms of IBD share many clinical and epidemiologic characteristics, suggesting that underlying causes may be similar. Indeed, more than occasionally, Crohn’s disease cannot be distinguished from UC on clinical grounds, yet the two diseases are distinct syndromes with divergent treatment and prognosis.
HISTORY OF CROHN’S DISEASE
Although the eponym Crohn’s disease has gained general acceptance in recent decades, clear clinicopathologic reports of the same process date back at least two centuries. Morgagni provided a description of intestinal inflammation characteristic of Crohn’s disease in 1761.1 Only after the identification of the tubercle bacillus by Koch in 1882 was it possible to describe persons with ileocecal disease similar to intestinal tuberculosis but lacking the organism. Such reports were provided by Fenwick (1889),1 Dalziel (1913),2 Weiner (1914), Moschcowitz and Wilensky (1923 and 1927), and Goldfarb and Suissman (1931).3 In 1932, the landmark publication of Crohn, Ginzburg, and Oppenheimer called attention to “terminal ileitis” as a distinct entity and chronic disease.4 This term was soon deemed unsuitable, however, when it became apparent that the disease process might involve the colon. Patients, too, misunderstood and were frightened by the “terminal” nature of their illness. The term “regional enteritis” embraced the focal nature of the process, but failed to incorporate knowledge of the possibility of disparate sites of involvement within the gastrointestinal tract, including the small and large bowel in combination5 and large bowel in isolation.6 The term “granulomatous enterocolitis” lost acceptance when it became clear that granulomas were not a sine qua non of the diagnosis. In the end, the name “Crohn’s disease” has been adopted to encompass the many clinical presentations of this pathologic entity. But for the alphabetic priority these authors chose, Crohn’s disease might well have been Ginzburg’s or Oppenheimer’s disease.
EPIDEMIOLOGY
Misclassification of disease is problematic. Historically, unidentified infections, later recognized by improved culture and diagnostic techniques, might have accounted for some portion of cases, particularly among persons with a single episode of disease. At times, differentiating Crohn’s disease from UC may be difficult, especially at the time of diagnosis, and before the passage of time has allowed distinctive disease characteristics to become manifest. Reassignment of a diagnosis of Crohn’s disease or UC may be as high as 9% in the first two years after diagnosis.7
Despite these methodologic limitations, distinct and reproducible geographic and temporal trends in incidence have been observed. In both North America and Europe, higher incidence rates have been noted in more northern latitudes. For example, age-adjusted annual incidence rates of 9 per 100,000 persons were reported in Olmsted County, Minnesota,8 and Copenhagen County, Denmark,9 and as high as 20 per 100,000 in Nova Scotia.10 Comparatively, estimates of incidence rates reached only 0.9 per 100,000 in Spain11 and 3.4 per 100,000 in Italy.12 A north-south gradient similar to that observed in Europe13 has been noted in the United States14 and even within the state of California itself, with estimated incidence rates of 7.0 and 3.6 per 100,000 in northern and southern California, respectively.15,16 In Asian countries, the incidence rate has remained low, with a mean estimated incidence of 0.5 per 100,000 persons in Korea17 and a similar incidence in Japan,18,19 whereas in Australia and New Zealand, incidence rates have ranged from 1.75 to 2.1 per 100,000.20,21 Crohn’s disease is thought to be extremely rare in much of South America and Africa22 with the exception of South Africa, where the most recent estimate of the incidence rate for the white population is 2.6 per 100,000; it is considerably lower among the nonwhite population.23
In some regions of the world where Crohn’s disease was rare, although still low compared with western countries, incidence is rising dramatically. For example, in Seoul, South Korea, the incidence increased from 0.05 per 100,000 in the early 1980s to 1.34 per 100,000 between 2001 and 2005.17 This trend has been seen throughout other regions as well.24–27 Estimates from less-affluent nations may be influenced by increasing access to health care, and therefore, genetic and environmental factors in these regions are difficult to disentangle.
Incidence rates have continued to rise in some regions, such as in Denmark,9,28 whereas in others they appear to be stabilizing. In Olmsted County, Minnesota, rates had been steadily increasing from approximately 3 per 100,000 (1954-1963) to nearly 8 per 100,000 (1964-1973), but since the late 1970s these rates have not changed significantly.8,29 Mortality trends for Crohn’s disease have followed a similar pattern, with rising mortality until the mid-1970s and stable rate since.30 Although improved diagnostic capabilities might have played some role in the rising incidence leading up to the mid-1970s, the fact that Crohn’s-related mortality was rising in parallel argues against the theory that rising Crohn’s diagnoses merely represented detection bias involving mild cases. Most recently, the prevalence of Crohn’s disease in the United States is estimated to be 201 per 100,000 adults and 43 per 100,000 in children, adolescents, and adults younger than 20 years.14
Studies throughout the world have shown a small excess risk of Crohn’s disease among women. Most reports show a female-to-male ratio in adult patients between unity and 1.3 : 1.10,14,28 In the pediatric population this is reversed, with more boys having Crohn’s disease than girls.14 This slight difference in risk in adult-onset disease may be explained by hormonal or life-style factors and stands in contrast to the nearly equal or even slight male predominance seen in UC.
Crohn’s disease is diagnosed most often among persons 15 to 30 years of age, although the age of diagnosis can range from early childhood throughout an entire lifespan. Population-based studies have shown the median age of diagnosis to be approximately 30 years.8,10 Conflicting information may be found regarding trends in the age of diagnosis. In Olmsted County, Minnesota, younger age groups have had a fairly stable incidence over the past 20 years, with rising rates in patients aged 60 and older.8,10 This trend of older-age diagnoses also was seen in population-based studies in Copenhagen, Denmark,31 and in Stockholm, Sweden,32 the median age of diagnosis increased from 25 years in 1960 to 1964 to 32 years in 1985 to 1989. These findings reflect diagnosis in a larger proportion of patients older than 60 years. Indeed, many, though not all,10 studies have shown a smaller second peak in incidence later in life, generally in the seventh decade.33 This second peak may be the result of ascertainment bias because of more frequent contact with medical care and more frequent evaluation of older patients. Differences in clinical presentation among younger and older patients suggest that distinct risk factors are operative at different ages at onset.34 The pathologic findings in young and old patients are not different, although some studies have identified a greater proportion of colonic and distal disease among older patients,33 compared with a predominance of ileocolonic disease in younger patients.35
ETIOLOGY AND PATHOGENESIS
INITIATING EVENTS
In light of the nature of the pathologic findings in Crohn’s disease (see later) and UC, it long has been clear that IBD represents a state of sustained immune response. The question arises as to whether this is an appropriate response to an unrecognized pathogen or an inappropriate response to an innocuous stimulus. Many infectious agents have been proposed as the cause of Crohn’s disease including chlamydia, Listeria monocytogenes, cell-wall–deficient Pseudomonas species, reovirus, and many others. Paramyxovirus (measles virus) has been implicated etiologically in Crohn’s disease as a cause of granulomatous vasculitis and microinfarcts of the intestine,36 but a proposed association between early measles vaccination and Crohn’s disease largely has been disproved.37 Another suggestion has been that the commensal flora, although normal in speciation, possess more subtle virulence factors, such as enteroadherence, that cause or contribute to IBD.38
Among the most enduring hypotheses is that Mycobacterium paratuberculosis is the causative agent of Crohn’s disease. This notion dates to Dalziel’s observation in 1913 that idiopathic granulomatous enterocolitis in humans is similar to Johne’s disease, a granulomatous bowel disease of ruminants caused by M. paratuberculosis.39 M. paratuberculosis is extremely fastidious in its culture requirements, and some proponents of this hypothesis have speculated that the presence of M. paratuberculosis as a spheroplast may have confounded efforts to confirm this hypothesis by culture of the organism; demonstrating it by immunohistochemistry, in situ hybridization, and polymerase chain reaction methodology; and empirical treatment with antimycobacterial antibiotics (see “Medical Therapy,” later). Most investigation in this area has been inconclusive, providing insufficient evidence to prove or reject the hypothesis.
In light of the diversity of substances and bacteria within the intestinal lumen, it is remarkable that the intestine is not perpetually inflamed. The presence of low-level physiologic inflammation within the healthy intestinal mucosa represents a state of preparedness to deal with potentially harmful agents. A more vigorous response would not be appropriate if directed toward the innocuous commensal flora of the intestine. Experiments in animal models of IBD suggest that in a genetically susceptible host, a classic pathogen is not necessary to cause IBD, but rather nonpathogenic commensal enteric flora are sufficient to induce an inappropriate chronic inflammatory response. In diverse models, animals raised under germ-free conditions show diminished or delayed expression of the IBD phenotype.40 On introduction of defined bacterial flora, however, the expected phenotype of bowel inflammation becomes manifest.40 Such models suggest that a diversity of genetic alterations, including those that affect intestinal barrier function and regulation of mucosal immunity, can result in intestinal inflammation. As in the animal models of IBD, evidence in patients with Crohn’s disease also points to an over-responsiveness of mucosal T cells to the enteric flora, manifest in part by the presence of antibodies against an array of bacterial antigens, including Escherichia coli outer membrane porin C (OmpC) and flagellin. Advances in the genetic bases of Crohn’s disease confirm that diverse bacterial agents are capable of fueling the inflammation of Crohn’s disease (see later).
GENETICS
The argument for a genetic predisposition to IBD begins with the observation that family members of affected persons are at greatly increased risk for developing IBD. The relative risk among first-degree relatives is 14 to 15 times higher than that of the general population.41 Roughly one of five patients with Crohn’s disease report having at least one affected relative. Many families have more than one affected member, and although there is a tendency within families for either UC or Crohn’s disease to be present exclusively, mixed kindreds also occur, suggesting the presence of some shared genetic traits as a basis for both diseases, as has recently been confirmed.
Ethnicity also plays a role. Eastern European (Ashkenazi) Jews are at a two- to four-fold higher risk of developing IBD than non-Jews of the same geographic location, and they are at greater risk of having multiple affected family members. Studies of monozygotic and dizygotic twins suggest that genetic composition is a more powerful determinant of disease for Crohn’s disease than for UC: The concordance rate among monozygotic twins is as high as 67% for Crohn’s disease but only 13% to 20% for UC. Most studies have suggested that concordance of disease location and disease behavior are higher than one would expect by chance.42 Finally, some subclinical markers of Crohn’s disease, including anti-OmpC antibodies, are more common among apparently healthy family members of Crohn’s disease probands than among the general population.43
Early studies of IBD genetics were limited by the slow speed of DNA sequencing and an incomplete understanding of the human genome’s structure. Studies of IBD genetics depended upon selecting a candidate gene based upon the then-current understanding of pathogenesis. With automated, rapid DNA sequencing, and mapping of the common genetic variants that occur in humans (the HapMap, or haplotype map), genome-wide association (GWA) studies became feasible. GWA studies can simultaneously explore many hundred thousands of genetic markers, providing a broad and unbiased approach to assessing the association of genomic loci to a specific disease without prior hypothesis about a candidate gene. GWA studies have been more successful in Crohn’s disease than in any other complex disease, and they have accelerated the pace of gene discovery, providing unexpected insights into pathogenesis.44 More than 30 genetic loci have been convincingly associated with Crohn’s disease in a large and powerful GWA study, confirming some known loci and discovering many loci that had not been described previously (Table 111-1).45 Thus far, genetic studies have highlighted three pathways of fundamental importance in the pathogenesis of Crohn’s disease.
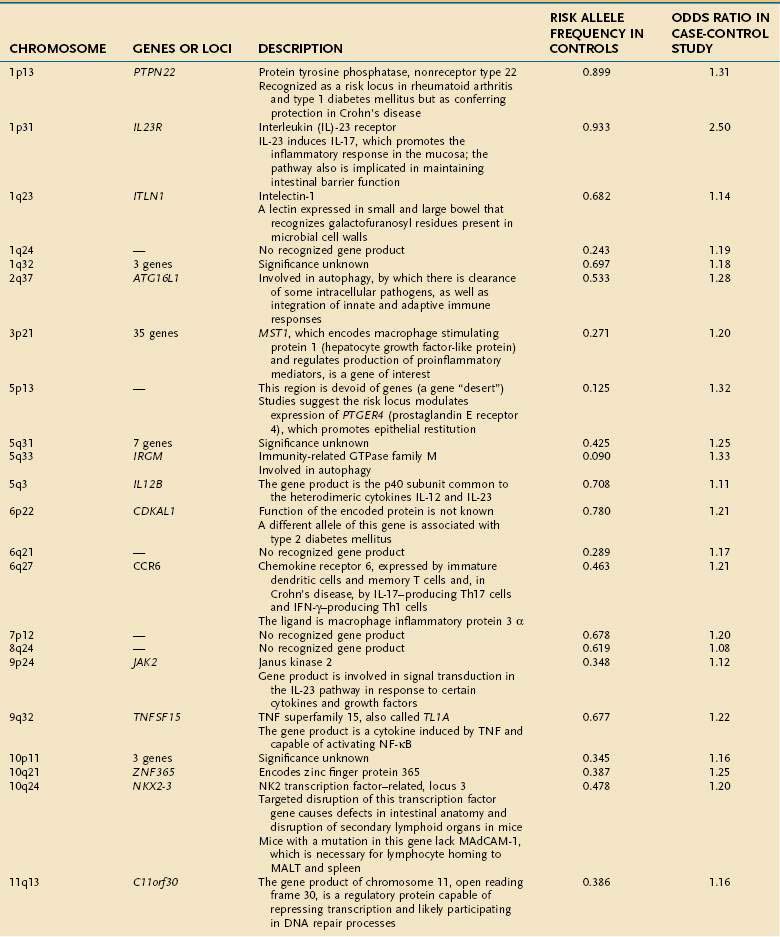
The first susceptibility locus for Crohn’s disease was identified in 2001 as the NOD2 (nucleotide-binding oligomerization domain 2) gene, also known as CARD15 (caspase-recruitment domain 15).46,47 The allelic variants most commonly associated with Crohn’s disease in European and American populations include one frameshift insertion leading to early truncation of the protein (Leu1007fsinsC) and two missense mutations (Arg702Trp, Gly908Arg). Carriage of disease-associated allelic variants on both chromosomes confers an odds ratio for Crohn’s disease of 17.1 (95% confidence interval [CI], 10.7-27.2), and heterozygous persons have an odds ratio of 2.5 (95% CI, 2.0-2.9) for the disease.48 Studies have associated genetic polymorphisms of NOD2/CARD15 with younger onset and ileal location of disease and increased likelihood of stricture formation.49–52 It has been estimated that as many as 20% to 30% of patients with Crohn’s disease bear abnormal NOD2/CARD15. Nevertheless, the penetrance of NOD2/CARD15 is estimated to be less than 1%53; that is, disease-related allelic variants of the gene may be found in a large number of persons who do not have Crohn’s disease; this strongly suggests that environmental factors, as yet incompletely elucidated, play a significant role in the expression of the Crohn’s disease phenotype (see later).
The discovery of the association of NOD2/CARD15 with Crohn’s disease has opened a remarkable window into the pathogenesis of Crohn’s disease. The gene product of NOD2/CARD15 is a cytosolic protein that functions as an intracellular sensor of bacteria. Specifically, the protein binds to muramyl dipeptide (MDP; MurNAc-L-Ala-D-isoGln), a component of bacterial peptidoglycan, found in Gram-positive and Gram-negative bacteria.54,55 The NOD2/CARD15 protein is expressed in monocytes and enterocytes, specifically in Paneth cells,56 which lie within the crypts and produce the endogenous antimicrobial peptides called defensins. The NOD2/CARD15 gene consists of two CARD domains, a nucleotide binding domain (NBD), and 10 leucine-rich repeats (LRR). NOD2/CARD15 variants associated with Crohn’s disease lie within the LRR and interfere with binding to MDP. In mononuclear cells, mutations in NOD2 result in decreased activation of nuclear factor (NF)-κB, whereas an excess of NF-κB expression is observed in tissue inflamed by Crohn’s disease. This apparent paradox has yet to be unraveled completely, but it is clear that defects in NOD2 impair antibacterial responses, particularly to oral exposure to pathogens. Notably, the production of β-defensins, which are antibacterial proteins produced by Paneth cells, is defective in Crohn’s patients with variant NOD2.57 These findings strongly implicate defects in innate immunity—the immediate and nonspecific immune responses to microbial infection—in a subset of patients with Crohn’s disease, with subsequent chronic activation of adaptive immunity, the antigen-specific responses mediated by antigen presenting cells (APCs) and T cells.
Subsequent discoveries have implicated defects in multiple genes in the autophagy pathway in the pathogenesis of Crohn’s disease.58,59 Autophagy is an ancient cellular process, highly conserved in evolution, by which segments of cytoplasm are isolated within a membrane and delivered to lysosomes by mechanisms that do not involve transport through endocytic or vacuolar sorting pathways. This unique process plays a role in cellular homeostasis by clearing proteins that are long-lived, misfolded, or aggregated, and by clearing apoptotic bodies, which might otherwise trigger inflammation and autoimmunity. Autophagy has been shown to contribute directly to innate immunity through direct killing of pathogens, activation of Toll-like receptors and Nodd-like receptors, and elaboration of immunomodulatory cytokines such as interferon (IFN)-γ. Autophagy also stands at the interface of innate and adaptive immune responses, delivering antigen to human leukocyte antigen (HLA) class II molecules in APCs for antigen-specific binding.59
GWA studies have identified variants that predispose to Crohn’s disease in at least two autophagy-related genes. The first, the autophagy-related 16-like 1 (ATG16L1) gene, was noted as having a disease-associated single nucleotide polymorphism (SNP) encoding an amino acid substitution in exon 8, resulting in a change from alanine to threonine58–60; this minor allele is protective against Crohn’s disease. ATG16L1 is expressed by intestinal epithelial cells, APCs, and various subsets of human T cells. The second autophagy gene associated with Crohn’s disease is the IRGM (immunity-related GTPase [guanosine triphosphatase] family member M) gene on chromosome 5q33.1.61 Careful study suggests that the disease-associated variants of this gene do not affect the amino acid sequence of its product, but they more likely alter its expression.61 IRGM appears to be important in resistance to intracellular pathogens such as mycobacteria, Listeria monocytogenes, and Toxoplasma gondii.59
A third pathway associated with Crohn’s disease is interleukin (IL)-23 and other gene products associated with this protein.62 IL-23 is a heterodimeric cytokine comprising two linked subunits (p19 and p40). IL-23 is produced by many cell types, including dendritic cells and macrophages, in response to diverse microbial signals. Naïve CD4+ T cells up-regulate IL-23 receptor when exposed to IL-6 and transforming growth factor (TGF)-β, completing an autocrine loop in the generation of Th17 T cells, effector T cells that produce IL-17.63,64 A rare variant of the IL23R gene leading to a glutamine at position 381 rather than an arginine is strongly protective for Crohn’s disease, with an odds ratio of 0.26 to 0.45; other, more common SNPs are associated with increased risk for Crohn’s disease and UC.65 In the same pathway, variants of the IL12B gene, encoding the p40 subunit common to IL-12 and IL-23, and of the JAK2 and STAT3 genes, with roles in IL23R signaling, as well as in Th17 differentiation in the case of STAT3, also have been associated with Crohn’s disease susceptibility.45 Together, these findings support the pivotal role of this pathway in maintaining mucosal homeostasis in the normal intestine. As the functional alterations associated with the many other identified genetic risk loci are elucidated, it is certain that new insights into the causes of Crohn’s disease will arise.
ENVIRONMENT
Although the greatest relative risk of Crohn’s disease is found among first-degree relatives of affected persons, particularly siblings of the proband, environmental factors also are important. As noted earlier, the rising incidence of Crohn’s disease over many decades highly suggests an environmental contribution to the expression of disease. Epidemiologic studies have examined numerous risk factors for Crohn’s disease. Most studies have found breast-feeding to be protective for IBD, presumably by playing a role in early programming of immune responses in the developing gastrointestinal tract. Occupations associated with outdoor physical labor are relatively under-represented among Crohn’s patients. Crohn’s disease has been associated with higher socioeconomic status,66 presumably because of relative underexposure to diverse environmental antigens in the course of childhood—the hygiene hypothesis as it relates to intestinal mucosal immunity in IBD. Many, but not all, studies have discerned an increased risk of Crohn’s disease among women who use oral contraceptives. Nonsteroidal anti-inflammatory drugs (NSAIDs) have been implicated not only in exacerbations of IBD but also as a potential precipitant of new cases, perhaps by increasing intestinal permeability. Increased intake of refined sugars and a paucity of fresh fruits and vegetables in the diet have been associated with the development of Crohn’s disease. It is conceivable that this observation may be confounded by exacerbation of symptoms in patients with mild disease because of increased dietary fiber intake and subsequent avoidance of these food items before diagnosis.
Smoking is one of the more notable environmental factors for IBD. UC is largely a disease of ex-smokers and nonsmokers, whereas Crohn’s disease is more prevalent among smokers. In addition, smokers have more surgery for their disease and a greater risk of relapse after resection. The reasons for the divergent effect of smoking on Crohn’s disease and UC are poorly understood, but they might include effects on intestinal permeability, cytokine production, and clotting in the microvasculature. More recently, studies have focused on the role of carbon monoxide in stimulating immunosuppressive effects mediated by heme oxygenase-1.67
Many patients report a correlation between disease exacerbations and stress. Although depression and anxiety are a common reaction to illness, Crohn’s disease has not been shown to be caused by stress or by an anxious personality. The mind-body connection between emotional states or stress and intestinal inflammation in IBD is slowly being revealed, however, and studies indicate that stress may be associated with risk of relapse in Crohn’s disease.68
ADAPTIVE IMMUNE RESPONSE AND INFLAMMATION
The interaction between effector T cells and APCs is critical to the pathogenesis of Crohn’s disease (Fig. 111-1). The antigens that perpetuate the inflammatory response are taken up by APC. Degradation of antigen within proteasomes results in presentation of an epitope in the context of major histocompatibility complex (MHC) class II. Interaction between MHC class II and the T-cell receptor (CD3) results in antigen-specific interaction between the macrophage and the CD4+ T cell. This event is necessary, but not sufficient, to activate the T cell. A second costimulatory signal is needed as well, because binding of CD3 to MHC class II without a costimulatory signal can result in anergy or apoptosis. Important costimulatory signals include binding of tumor necrosis factor (TNF) to TNF receptor, CD40 to CD40 ligand, and B7 to CD28. Activation of T cells leads to production of IL-2, an important growth factor for T cells.
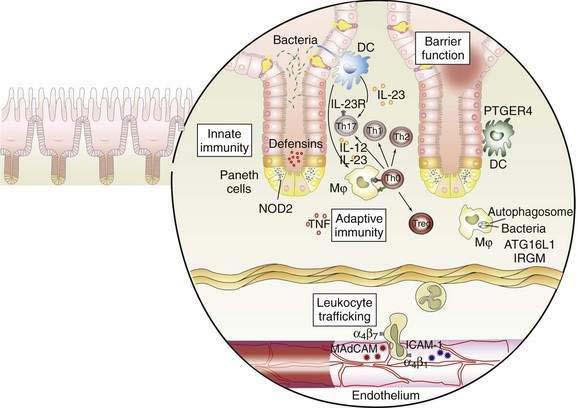
As noted earlier, the p40 subunit is common to IL-12 and IL-23, each of which, in turn, is critical in shaping the Th1 and Th17 responses that characterize Crohn’s disease. In addition to IL-23, the presence of TGF-β and IL-6 facilitate differentiation of naïve T cells into pathogenic Th17 cells.63 Activated APCs further shape and amplify the immune response by producing the T cell growth factor IL-2 and the proinflammatory cytokines IL-1 and TNF. Within mononuclear cells, the key nuclear transcription factor is NF-κB, which regulates the transcription of IL-1, IL-6, IL-8, TNF, and other peptides central to the inflammatory response.69
NF-κB is regulated tightly within the cell. In the inactive state, NF-κB is held in the cytoplasm, bound to inhibitory κBα. During cell activation after receptor binding, various kinases phosphorylate inhibitory κBα, thereby leading to its degradation. NF-κB is then released, permitting translocation to the nucleus, where it binds to the promoter regions of numerous genes that support the inflammatory response. Such genes include those that encode proinflammatory cytokines such as TNF, adhesion molecules, and chemokines.69
Expression of adhesion molecules is critical to amplify the immune response, because the resident populations of granulocytes and mononuclear cells alone do not account for the vigorous inflammatory reaction characterizing IBD. Adhesion molecules on the leukocyte surface and their ligands on the endothelium of venules in the lamina propria interact in a coordinated multistep process that permits trafficking of inflammatory cells into the mucosa. First, a weak interaction between selectins on the leukocyte surface and the endothelium leads to rolling of the leukocytes along the endothelium. Second, in the presence of chemokines such as IL-8, activation occurs, and integrins are expressed on the leukocyte surface. Third, interactions between leukocyte integrins and immunoglobulin-like cellular adhesion molecules on the endothelial surface lead to spreading of the cell and diapedesis.70 Specificity is conferred by the presence of tissue-specific cellular adhesion molecules. The integrins α4β7 and αEβ7 are of special importance in IBD, because the corresponding ligands—mucosal addressin cellular adhesion molecule and E-cadherin—are intestine specific. Mucosal addressin cellular adhesion molecule is expressed constitutively on the endothelium of venules in the lamina propria,70 whereas binding of αEβ7 on intestinal lymphocytes to E-cadherin on intestinal epithelium permits localization of intraepithelial lymphocytes. Antibodies to the α4 subunit of integrin have proved to be therapeutic in Crohn’s disease.71
Once recruited to the lamina propria, mononuclear cells and granulocytes elaborate a variety of injurious and proinflammatory substances that ultimately cause tissue destruction. These substances include prostaglandins, reactive oxygen metabolites, nitric oxide, leukotrienes, and proteases. Collagenase and matrix metalloproteinases play a pivotal role in the tissue destruction seen in IBD.72 Counterbalancing these destructive substances are other substances that promote epithelial restitution and repair, including IL-11, trefoil peptides, and growth factors such as epidermal growth factor and keratinocyte growth factor.
PATHOLOGY
Focal intestinal inflammation is the hallmark pathologic finding in Crohn’s disease. This tendency for focal inflammation is evident in focal crypt inflammation, focal areas of marked chronic inflammation, the presence of aphthae and ulcers on a background of little or no chronic inflammation, and the interspersing of segments of involved bowel with segments of uninvolved bowel. Even within a single biopsy specimen one can see a pronounced variability in the degree of inflammation. The presence of focally enhanced gastritis, characterized by a focal perifoveolar or periglandular lymphomonocytic infiltrate, is a common finding that occurs in 43% of unselected patients with Crohn’s disease.73 This finding underscores the focal nature of the inflammation, despite the strong potential for inflammation to occur anywhere along the entire longitudinal axis of the gastrointestinal tract. To a certain extent, the nature of the findings and the depth of inflammatory changes depend on the chronicity of the inflammation.
EARLY FINDINGS
Because of the variable and often long delay between the onset of the disease process and its diagnosis, it rarely is possible to observe the evolution of pathology from the earliest events. Studies of recurrent Crohn’s disease after ileal resection have offered a window into the sequence of pathologic changes in the disease.74









