Fig. 57.1
Biliary development and the ductal plate malformation (modified from [1]). a At 14 weeks of gestation, hepatoblasts in contact with portal vein mesenchyme adopt a biliary epithelial fate. b At 17 weeks of gestation, biliary layer duplicates and focal dilatation within the layer leads to bile ducts formation. c At birth, bile duct development is nearly completed, and the bilayer has involuted. Liver cells are either hepatocytes or hepatoblasts. d Ductal plate malformation: abnormal ductal plate remodeling leads to saccular, biliary dilatations
The Ductal Plate
The ductal plate is the embryonic precursor of the intrahepatic bile ducts. It is first detected by 6–7 gestational weeks and consists of a double layer of cuboidal, biliary-type epithelium that surrounds the developing portal veins. This layer develops from the transformation of hepatoblasts and yields an anastomosing tubular network of biliary ducts through a process called ductal plate remodeling (Fig. 57.1) [4–7].
Ductal plate remodeling begins by 11 weeks of gestation, and in normal conditions it is complete in early postnatal life. It is a process of negative selection of nonfunctional ductal plate elements which involute in a centripetal fashion from the hilum toward the liver periphery [4, 6, 8]. The remodeling phase is characterized by a high rate of mesenchymal proliferation, resulting in the separation of the developing bile ducts from the liver parenchyma (Fig. 57.1) [4, 8]. Meanwhile, hepatocytes and intrahepatic biliary epithelial cells undergo apoptosis at a higher rate than normal under the control of apoptosis-related proteins: C-myc protein and fas antigen stimulate cell-death while Bcl-2 protein inhibits apoptosis [5]. It is possible that an imbalance between proliferation and apoptosis impedes normal ductal plate remodeling, thereby yielding the DPM [4, 5]. Remodeling is under the control of a number of genetic and molecular factors that make it vulnerable to maldevelopment. Among these factors are signaling molecules from the portal venous mesenchyme, from adjacent hepatoblasts, from involuting bile ducts, and hormones such as estrogens.
The Ductal Plate Malformation
The term “ductal plate malformation” was first used by Mogens Jorgen Jorgensen in 1977 to describe an unusual histologic configuration of the liver of children with polycystic disease. He first described extensive fibrosis and increased number of bile ducts within the fibrotic regions, now recognized as the cardinal features of the DPM [9]. It is now understood that the DPM occurs when excessive ductal plate remnants fail to disappear. What is not understood is why the size of the bile ducts affected by the DPM varies between diseases: Small ducts are affected in CHF while larger ducts are dysmorphic in CD. Figure 57.2 [10] illustrates the DPM and related diseases along what might be considered a centrifugal axis from the hilum to the subcapsular region. In the case of complete lack of remodeling of the ductal plate, the DPM appears like a circular lumen surrounding a central fibrovascular axis. Alternatively, incomplete remodeling results either in a ring of interrupted ducts around a fibrovascular axis or a polypoid projection of biliary epithelium in a dilated duct [10, 11] .
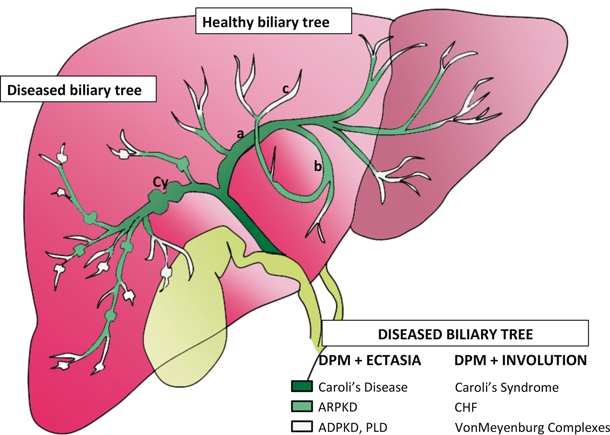
Fig. 57.2
Schematic of intrahepatic biliary tree and level of DPM [10]. For reasons still unknown, defects in ductal plate remodeling impact the biliary tree at different levels. In addition, the remodeling defect yields one of two phenotypes: predominantly fibrotic lesions or dilated biliary structures. Caroli’s syndrome is an example of how the two phenotypes might overlap. DPM ductal plate malformation, CHF congenital hepatic fibrosis, ARPKD autosomal recessive polycystic kidney disease, ADPKD autosomal dominant polycystic kidney disease, PLD polycystic liver disease
There are two groups of diseases in which the DPM is a dominant feature. The first is characterized by inflammation and destruction of intrahepatic bile ducts with some degree of fibrosis, as seen in biliary atresia. The second is the pattern typically seen in fibrocystic diseases: extensive fibrosis with ectasia and hyperplasia of intrahepatic bile ducts (Fig. 57.2) [7]. Our purpose is to focus on the latter group.
Cavernous Transformation of the Portal Vein
The DPM is often associated with abnormalities in portal vein branching. During normal development, the portal vein invades the early liver parenchyma in a centrifugal branching pattern from the hilum to the periphery. This branching pattern determines the lobular architecture of the liver. Therefore, it follows that alterations of vascular development and intrahepatic bile ducts go hand in hand [7, 12, 13]. In the case of DPM, branching is abnormal, thereby leading to the “pollard willow formation,” characterized by clusters of unseparated venous tracts [7, 10, 12]. Extrahepatic cavernous transformation of the portal vein occurs in 50–70 % of patients with CHF and CD and contributes to portal hypertension, one of the main clinical features of CHF patients [12, 13]. Congenital absence of the portal vein has rarely been reported in association with CHF [14, 15].
Fibrosis
CHF is a developmental disorder due to DPM of the small, interlobular bile ducts, during which persistence of embryonic remnants stimulates the development of fibrosis. Portal fibrosis is the major histological finding in CHF. The development of fibrosis is a multifactorial process involving hepatic stellate cells (HSC), myofibroblasts, and macrophages. Known molecular mediators include transforming growth factor-β1 (TGF-β1) and connective tissue growth factor (CTGF) [16]. CTGF is known to promote fibrosis [16]. In CHF, CTGF is retained in the extracellular matrix (ECM), thereby maintaining a local stimulus for fibrosis. How fibrosis develops in ciliopathies is further explained in the section below on animal models.
HSC constitute approximately 5–8 % of liver cells. They are located in the space of Disse, between sinusoidal endothelial cells and liver parenchymal cells [17, 18]. Activated stellate cells express tissue inhibitors of metalloproteinases 1 and 2 (TIMP 1 and 2) [19–21]. These factors have two roles in fibrogenesis: they exert a direct inhibitory action on metalloproteinases which leads to ECM accumulation, and they confer antiapoptotic proliferative signals to activated stellate cells [19, 22]. Finally, stellate cells respond to nearby injury by transforming into myofibroblasts [17] which secrete ECM components leading to scarring [17, 18].
Myofibroblasts are only present in the injured liver, and are classically believed to derive from portal fibroblasts or HSC. They are responsible for producing the ECM which leads to fibrosis [18, 23]. In CHF, parenchymal fibrosis is minimal compared to portal fibrosis. Therefore, it is possible that, in CHF, myofibroblasts derive from periductal fibroblasts rather than from parenchymal stellate cells [16].
Finally, macrophages have also been recognized as important regulators of liver fibrosis. They are located close to myofibroblasts and interact with the latter and with HSCs [24–27]. They also act independently, by producing their own metalloproteinases and TIMP. They contribute to the phagocytosis of cellular debris and to the secretion of cytokines and immunoregulatory mediators [24, 27, 28].
In summary, portal fibrosis is a major feature of CHF. In response to abnormal ductal plate remodeling, portal fibroblasts transform into myofibroblasts and interact with macrophages which secrete the inflammatory cytokines that trigger fibrosis. At this time, however, the underlying molecular and cellular mechanisms are not clear.
Ciliopathies
Abnormal primary cilia are now understood to be one of the main mechanisms causing DPM. The term ciliopathies refers to a heterogeneous group of disorders involving anomalies in primary cilia. Primary cilia are highly conserved organelles used to detect various extracellular stimuli [29]. Although their existence has been known since the nineteenth century, their role in disease is still incompletely understood.
In the liver, cholangiocytes are the only cells with primary cilia [30, 31]. Normal ciliary function is necessary for biliary development. Cholangiocyte cilia are important receptors of mechanical and osmotic signals. They respond by modulating intracellular levels of cyclic adenosine monophosphate (cAMP) and calcium in response to bile flow. This function depends on polycystin 1 and 2 (PC-1 and PC-2) that form a functional complex. PC-1 is a ciliary protein with a large extracellular domain that senses cilia bending. It then transfers this signal to PC-2 which triggers a rise in intracellular calcium which in turn regulates cellular proliferation. PC-1 exerts a regulatory role on growth, such that when its function is impaired or its level of expression diminished, the break is lifted and excess proliferation ensues [32]. Cholangiocyte cilia also detect osmotic changes in bile via the transient receptor potential cation channel subfamily V member 4 (TRPV4) protein which participates in the regulation of bicarbonate-dependent bile secretion (Fig. 57.3) [30, 31, 33–36].
Structural and functional defects of primary cilia result in decreased intracellular calcium and increased cAMP, causing cholangiocyte hyperproliferation and abnormal cell–matrix interactions [30, 34, 37–39]. Excessive cholangiocyte proliferation leads to the development of cysts. Estrogens and insulin-like growth factor 1 (IGF-1) also contribute to cholangiocyte proliferation, and cystic cells have been shown to overexpress estrogen and IGF-1 receptors [38, 40]. Furthermore, cell cycle dysregulation also contributes to cholangiocyte proliferation [38]. Finally, cystic cholangiocytes also display an abnormal ion transporter pattern leading to increased fluid secretion in the cysts [38, 41]. As summarized in Fig. 57.3, cyst expansion in ciliopathies is related to structural and functional defects of the cilia, leading to cholangiocyte proliferation and cyst formation. This process is enhanced by other factors, such as abnormal cell–matrix interactions, cell cycle dysregulation, overexpression of estrogen and IGF-1 and increased fluid secretion in the cysts.
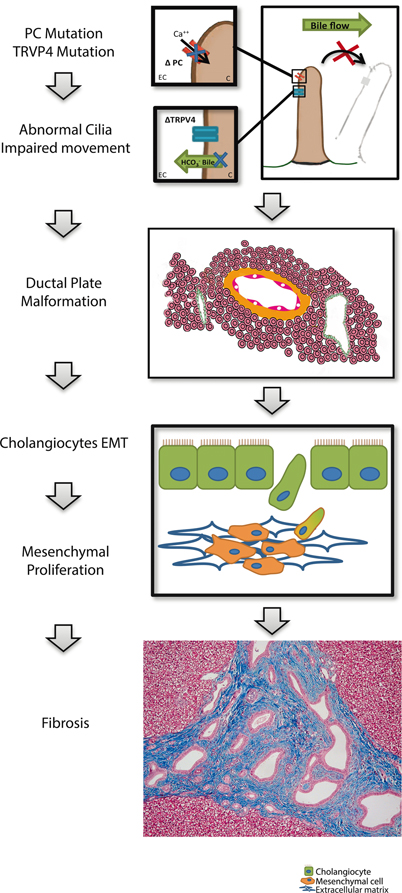
Fig. 57.3
Schematic description of molecular and cellular mechanisms in the hepatic manifestations of ciliopathies. Mutations in polycystin (PC) lead to impaired calcium transport into cilia, and thus impaired movement. In case of TRPV4 mutation, osmotic changes in bile cannot be sensed, and a dysregulation of bicarbonate-dependant bile secretion occurs. Structural and functional defects of the cilia lead to ductal plate malformation. Cholangiocytes undergo an epithelial to mesenchymal transition and proliferate in the mesenchyme contributing to fibrosis. PC polycystin, TRPV4 transient receptor potential cation channel subfamily V member 4, EC extracellular, C cytosol, EMT epithelial-to-mesenchymal transition
Evidence suggests that biliary precursors are vulnerable to second hits that can suffice to impair remodeling, for example, somatic mutations occurring during development [42, 43]. A mutation in a single allele of a developmentally key transcription factor such as hepatocyte nuclear factor-6 (HNF6) may lead to impaired ciliogenesis and thus cyst development. This can be a very early event that precedes the formation of the ductal plate by days or weeks and may involve genetic modification leading to the loss of heterozygosity, but may also involve other cellular events such as glycosylation and posttranslational modification leading to faulty transcription factor binding [44]. Among other developmental steps prone to anomalies the local growth-factor milieu is another variable in a complex web of events. It is probably because the number of genes and signaling pathways are so numerous and the possible combinations of developmental accidents almost infinite, that biliary development is so vulnerable to malformations and that there is so much overlap between phenotypes among the ciliopathies and fibrocystic diseases. To help make sense of this wide range of phenotypes, it has been suggested that DPMs can be categorized in one of three ways: perturbations in differentiation of biliary precursors, impaired maturation of radially asymmetrical primitive ductules and abnormal (excessive) ductular expansion.
Animal Models of Ciliopathies
Much of what is understood today about cystogenesis, fibrosis, and ciliopathies is through animal studies. Although different parameters have been identified leading to cholangiocyte proliferation and cyst development, until recently there were no clear mechanistic studies to explain the link between cyst formation and fibrosis. Recent studies in rats have shown that abnormal cystic cholangiocytes can undergo an epithelial to mesenchymal transition and secrete ECM molecules contributing to the development of fibrosis (Fig. 57.3) [45].
The PCK rat carries a splicing mutation in polycystic kidney and hepatic disease 1 gene (Pkhd1) an ortholog of polycystic kidney and hepatic disease 1 (PKHD1). Defects in ciliary structure of cholangiocytes lead to abnormalities of biliary tree differentiation and bile duct dilatation, mimicking CD. Besides the histological findings described above, macroscopic findings include cholangitis with intestinal metaplasia and hepatic fibrosis [28, 45, 46]. Therefore, this animal model will likely be pivotal in understanding the disease process and developing novel therapies.
Clinical Features
CHF and CD/CS are ciliopathies associated with ductal plate malformation . The liver findings are rarely an isolated finding. Rather, they are most often associated with polycystic kidney disease (PKD), part of the wider family of ciliopathies. When the phenotype combines hepatic and renal manifestations, these conditions are also referred to as the hepatorenal fibrocystic disorders (HRFCD).
CHF usually presents between 5 and 7 years of age. Familial cases exist, highlighting the importance of a thorough family history [47–50]. CHF can present in one of four ways [33, 50–52]. The most frequent presentation is portal hypertension with firm hepatomegaly and splenomegaly [47, 48, 50, 53, 54]. Thrombocytopenia and hypersplenism are often present. Occasionally anemia reveals occult variceal bleeding. The presenting complaint is often an acute gastrointestinal bleed in a previously healthy child. In 30–70 % of patients who present within the first decade of life, recurrent hematemesis or melena caused by variceal bleeding is frequent, whereas ascites is uncommon [52]. In most patients without an associated syndrome, growth and development are normal. Serum aminotransferase- and bilirubin levels are generally within the normal range at diagnosis and remain normal throughout follow-up. No progression to liver failure has been reported [49]. Complications of portal hypertension such as hepatopulmonary syndrome and porto-pulmonary hypertension have been described [49, 52].
The second presentation, frequently associated with CD/CS, is recurrent bacterial cholangitis. Symptoms include intermittent abdominal pain and pruritus from chronic cholestasis . In this case, serum levels of gamma-glutamyl transferase (gGT), alkaline phosphatase, and bilirubin are elevated. Bacterial cholangitis can progress to septicemia and lead to the development of hepatic abscesses. Recurrent cholangitis may lead to liver failure and failure to thrive [52].
Occasionally, patients can present with both portal hypertension and cholangitis. Finally, the disease can be latent and revealed by an incidental, clinical, biological, or radiological finding such as hepatomegaly or cytopenias [55–57]. When CHF or CD is associated with other extrahepatic signs, clinicians should think of associated syndromes, which are described in the following section.
In the long run, CD and CS are associated with an increased risk of cholangiocarcinoma, due to bile stasis and chronic inflammation. Cholangiocarcinoma, which can be the presenting feature, has been reported to occur in 5–10 % of patients with CD and CS. It has also been reported in patients with isolated CHF, albeit rarely. The average age at appearance is > 40 years (range of 33–75 years), and the risk increases with age. It occurs more often in the monolobar form of the disease, and has very poor 1-year survival rates [58–61].
Associated Syndromes and Ciliopathies
CHF and CD are often features of a multisystem disorder. Ciliopathies with a prevalence of more than 1:150,000 are described below and ciliopathies with low prevalence are outlined in Table 57.1 [62–73].
Disease | Phenotype | Prevalence | Gene abbreviation | Transmission | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
– | Retinal | Mental retardation | Cerebellar or cerebral | Cardiac | Lung | Pancreas | Liver | Kidney | Polydactyly | Infertility | Situs inversus | Other | – | – | – |
Autosomic recessive polycystic disease | – | – | – | – | – | – | X | X | – | – | – | – | 1:10,000 to 1:40,000 | PKHD1 | AR |
Autosomic dominant polycystic disease | – | – | – | – | – | – | X | X | – | – | – | – | 1:1000 | PKD1, PKD2 | AD |
Nephronophtisis | X | X | – | X | X | X | X | X | – | – | X | – | 1:100,000 | NPHP1, INVS, NPHP3, NPHP4, IQCB1, CEP290,GLIS2, RPGRIP1L, NEK, SDCCAG8, TMEM67, TTC21B, WDR19, ZNF423, CEP164, ANKS6, XPNPEP3 | AR |
Meckel Gruber | X | – | X | – | X | – | X | X | X | X | X | – | 1:140,000 | MKS1, TMEM67, CEP290, TMEM216, TCTN2 | AR |
COACH syndrome | X | – | X | – | – | – | X | – | – | – | – | – | < 1:1,000,000 | CC2D2A, TMEM67, RPGRIP1L | AR |
Joubert syndrome | X | X | X | – | – | – | – | X | X | X | X | – | – | JBTS1 to JBTS13 | – |
Bardet-Biedl [62] | X | X | – | X | X | – | X | X | X | X | X | – | 1:100,000 | BBS1, BBS2, ARL6, BBS4, BBS5, MKKS, BBS7, TTC8, BBS9, BBS10, TRIM32, BBS12, MKS1, CEP290 | AR |
Jeune syndrome [63] | – | – | – | – | – | – | X | X | X | – | – | Pelvic bone defect, thoracic defect | Rare | IFT80, DYNC2H1, TTC21B, WDR19 | AR |
Oral facial digital syndrome [64] | – | – | X | – | – | – | X | X | X | – | – | Cranio facial defect | 1:100,000 | OFD1 | XD |
Renal-hepatic-pancreatic dysplasia = Ivemark syndrome [65] | – | – | – | X | – | X | X | X | – | – | – | – | Rare | GDF1 | AR |
Ellis van Creveld syndrome [66] | – | – | – | X | – | – | – | – | X | – | – | Thoracic defect | Rare Increased in Amish people | LBN, EVC | AR |
Alstrom syndrome [67] | X | – | – | X | – | – | X | X | – | – | – | – | Rare | ALMS1 | AR |
Leber congenital amaurosis [68] | X | X | – | – | – | – | – | – | – | – | – | – | Rare | CEP 290, NPHP6, LCA 5, TULP I; REGRIP 1 | AR |
Senior Løken syndrome [69] | X | – | – | – | – | – | X | X | – | – | X | – | Rare | CEP 29, NPH1, NPHP, NPHP4, NPHP5 | AR |
Mainzer-Saldino syndrome [70] | X | – | X | – | – | – | X | X | – | – | – | Phalangeal, cone-shaped epiphyses | Rare | IFT140 | AR |
– | – | X | – | – | – | X | – | – | – | – | Enteropathy, coagulopathy | Rare | MPI | AR | |
Autosomal Recessive Polycystic Kidney Disease
Autosomal recessive polycystic kidney disease (ARPKD) is the most frequent ciliopathy in childhood, affecting 1:20,000 live births. It is characterized by progressive cystic degeneration of the kidneys and CHF. As its name suggests, transmission is recessive. All patients diagnosed with ARPKD have CHF from birth on microscopic examination, but only 50 % have hepatomegaly. Most of the patients are born with nephromegaly from dilated collecting ducts which can present as a palpable flank mass. Thirty percent of patients die at birth from pulmonary hypoplasia (Potter’s syndrome). There is a marked variability of phenotypes even among siblings. The course and the severity of renal disease and liver disease are unrelated. In the infant or neonate, presentation is usually renal, while CHF is typically asymptomatic early in life. Renal disease can present as a simple urinary concentration defect or more often as severe cystic kidney disease leading to hypertension and renal failure. End-stage renal disease affects about 30 % of children under 10 years. For neonates surviving the first month of life, the 5-year survival rate is about 90 %. Children who present later in life will show one of the CHF presentations described earlier. Diagnosis is clinical and by ultrasonography (US). Kidneys appear hyperechoic on ultrasound, and the liver is either normal or hyperechoic. Rarely, macroscopic liver cysts are visible on ultrasound. Annual follow-up should include a full blood count and ultrasound. MR cholangiography should be performed once to understand biliary morphology. Ultrasound examination of the parents’ kidneys is important because autosomal dominant disease can mimick the recessive form in neonates. Management is supportive, both for the renal and liver complications. Most patients presenting as neonates will require kidney transplantation before 20 years of age. Improvement in the management of renal complications has led to an increased awareness of liver-related morbidity and mortality. Liver-related mortality following kidney transplantation has been reported. Liver transplantation is performed in about 3 % of cases of ARPKD, usually during adolescence. Combined liver and kidney transplantation are indicated in renal failure with recurrent cholangitis or refractory complications of portal hypertension [74–79] .
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


