Congenital Abnormalities
Intestinal Malpositions
Intestinal malpositions include disorders of malrotation, malfixation, reversed rotation, incomplete rotations, fixation abnormalities, and situs inversus. Intestinal malrotations or nonrotations (Fig. 6.44) result from disordered or interrupted embryonic intestinal counterclockwise rotations around the superior mesenteric artery. The normal rotations and fixations are either incomplete or occur out of order. Nonrotations result from an interruption early in the rotation, so that the duodenal–jejunal loop remains on the right side of the abdomen. The cecal position varies, but it usually lies in the upper abdomen on the left side (Fig. 6.44). Incomplete rotations reflect interruptions occurring after the duodenal–jejunal loop has partially rotated around the superior mesenteric artery. Variants of classic rotation also occur. These include reversed duodenal and colonic rotation and reversed duodenal rotation and normal colonic rotation. In reversed rotation, the dorsal and ventral loops rotate to the left rather than to the right. The cecum lies in the right iliac fossa but the small bowel lies superficial to the transverse colon (Fig. 6.44), often herniating into the right colonic mesentery (104). The small bowel may also fail to rotate fully. As the intestine returns to the abdominal cavity, the small intestine rotates 180 degrees but fails to continue rotating to the normal point of fixation at the ligament of Treitz. Considerable variations exist in the degree of rotation beyond 180 degrees. A short small intestinal and mesenteric segment often becomes fixed to the retroperitoneum along a line generally confined to the right upper quadrant. This line of fixation, rather than anchoring the small bowel securely, becomes a new point of rotation, substituting for the superior mesenteric artery as a rotation point. Fibrous bands or adhesions form between the bowel and other abdominal structures in an attempt to secure the mobile bowel (Fig. 6.45). These commonly cross and compress the duodenum, obstructing it. Rotational abnormalities also complicate developmental defects when the intestine occupies an extra-abdominal position, as occurs in congenital diaphragmatic hernias or in abdominal wall defects (105).
Intestinal malrotations affect approximately 1 in 6,000 live births (106). Three percent of patients have associated abnormalities (Table 6.1). Patients present with signs and symptoms
of duodenal obstruction, intermittent volvulus, or acute life-threatening midgut volvulus characterized by bilious vomiting, abdominal distention, rectal bleeding, or intussusception. Infants may also develop malabsorption with steatorrhea and protein-losing enteropathy resulting from mesenteric lymphatic obstruction. Adults with malrotations often have a lifetime history of nonspecific abdominal complaints, including acute symptoms when they were children (107).
of duodenal obstruction, intermittent volvulus, or acute life-threatening midgut volvulus characterized by bilious vomiting, abdominal distention, rectal bleeding, or intussusception. Infants may also develop malabsorption with steatorrhea and protein-losing enteropathy resulting from mesenteric lymphatic obstruction. Adults with malrotations often have a lifetime history of nonspecific abdominal complaints, including acute symptoms when they were children (107).
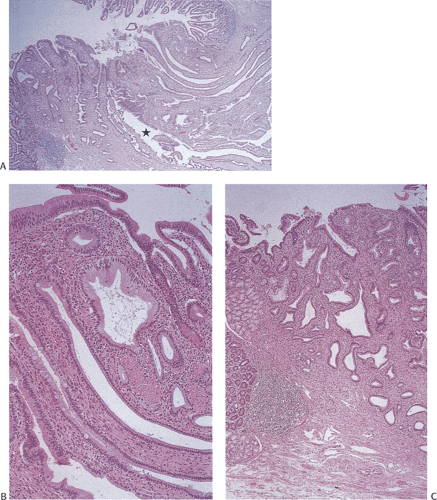 FIG. 6.43. Ampulla of Vater. A: Low magnification demonstrating the presence of numerous branching glands that enter at the area of the ampulla. A cross section of a larger duct is indicated by the star. B: Derives from the area to the right of the star in A. C: Higher magnification from the area to the left of the star. |
Malrotations exhibit obvious intestinal misplacement within the abdominal cavity. The intestines often lie to one side, appearing as a large mass of nonrotated bowel (Fig. 6.44). The entire bowel, from the duodenum to splenic flexure, remains unanchored and supported by a single mesentery with a very narrow base, predisposing the intestines to torsion and volvulus.
Situs Inversus
In situs inversus, the organs lie in mirror image locations of their normal positions. When complete, it affects both thoracic and abdominal organs. When incomplete, it affects only the abdominal organs. Limited situs inversus affects only the stomach and duodenum. Situs inversus affects 1 in 1,400 live births and it forms part of Kartagener syndrome.
Many children with situs inversus and a neural tube defect have mothers with insulin-dependent diabetes mellitus. Partial
situs inversus usually associates with other malformations, including asplenia, duodenal stenosis, and cardiac defects. Major associated gastrointestinal anomalies include annular pancreas, midgut volvulus, duodenal atresia, and mucosal duodenal diaphragms. Situs inversus does not change organ function or histology. One group of patients that may have a somewhat worse prognosis than others are those with Kartagener syndrome. These patients have abnormal cilia and as a result they produce thick, tenacious bronchial and sinus secretions that lead to chronic sinusitis and bronchiectasis.
situs inversus usually associates with other malformations, including asplenia, duodenal stenosis, and cardiac defects. Major associated gastrointestinal anomalies include annular pancreas, midgut volvulus, duodenal atresia, and mucosal duodenal diaphragms. Situs inversus does not change organ function or histology. One group of patients that may have a somewhat worse prognosis than others are those with Kartagener syndrome. These patients have abnormal cilia and as a result they produce thick, tenacious bronchial and sinus secretions that lead to chronic sinusitis and bronchiectasis.
TABLE 6.1 Abnormalities Associated with Small Intestinal Malrotation | |
|---|---|
|
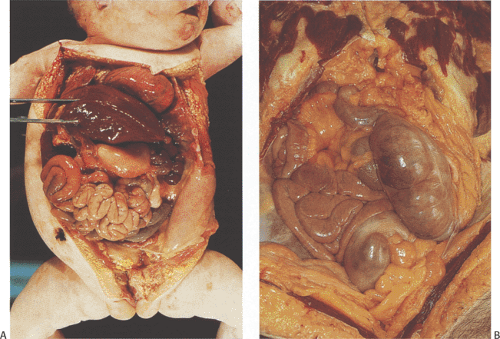 FIG. 6.44. Malrotation. A: The small intestines lie matted together in the lower abdomen, pushing the colon to the right side. B: A mass of unrotated small intestine lies on the right side of the abdominal cavity. The cecum lies on the left side. |
Omphaloceles
Omphaloceles consist of an external mass of abdominal contents covered by a variably translucent peritoneal and/or amniotic membrane. They result from the failure of the anterior abdominal wall to form completely during fetal development combined with the failure of the abdominal viscera to return to the abdomen at the end of the 10th fetal week. Omphaloceles affect 2.52 per 60,000 to 2 per 3,000 live births. Significant heterogeneity exists in the prevalence rates among different geographic regions, with especially high prevalence rates occurring throughout the British Isles. In the United Kingdom and Ireland, there is a tendency for omphaloceles to associate with anencephaly and spina bifida. The male-to-female incidence is 3:1; however, a significant female excess exists among the cases of omphalocele associated with neural tube defects. Up to 54% of infants with omphaloceles have associated anomalies (Table 6.2) compared with only 5% of those with gastroschisis (108,109). Many fetuses with an omphalocele have an abnormal karyotype. Chromosomal anomalies are particularly common in omphaloceles that do not contain the liver (110).
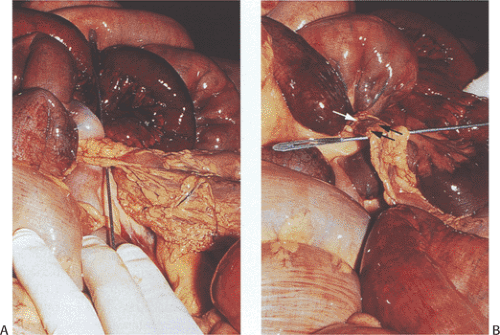 FIG. 6.45. Intestinal bands. Several views of the same intestinal band and its consequences. A: The mesenteries have herniated through a mesenteric defect underlying the band (probe). The surrounding bowel loops appear ischemic and erythematous secondary to a volvulus that occurred around the band. B: Further dissection shows the band elevated by the probe. A loop of dusky bowel is twisted around it (arrows). |
The rare OEIS (omphalocele, cloacal exstrophy, imperforate anus, spinal defects) complex affects 1 per 200,000 to 400,000 pregnancies. OEIS occurs sporadically or it affects twins or siblings from separate pregnancies, suggesting that some cases have a genetic basis. Some patients with omphalocele and the Miller-Dieker syndrome have a deletion at 17p13.3, suggesting that a gene in this region plays a major role in lateral fold closure or the return of the midgut from the body stalk to the abdomen. Trisomy 18 affects some patients. OEIS associates with a history of maternal diabetes mellitus or hydantoin or diazepam administration (111,112).
TABLE 6.2 Defects Associated with Omphaloceles | |
|---|---|
|
The OEIS complex arises from a single localized mesodermal defect early in development that contributes to infraumbilical mesenchymal, cloacal septum, and caudal vertebral abnormalities. Four somatic folds (a cephalic fold, two lateral folds, and a caudal fold) define the anterior thoracic and abdominal walls during the third fetal week. These folds migrate centrally to fuse at the umbilical ring, usually by the 18th gestational week. Arrested fold migration or development results in anterior wall defects and a widening of the umbilical ring.
The average birth weight and gestational age of infants with omphalocele is low. The amniotic membrane and peritoneum protect the developing intestinal loops from the damaging effects of exposure to amniotic fluid seen in gastroschisis. Omphaloceles range in size from only a few centimeters to lesions involving almost the entire anterior abdominal wall (Fig. 6.46). Abdominal viscera are present within a sac that initially is moist and transparent but with time becomes dry, fibrotic, opaque, friable, and prone to rupture with secondary evisceration. One often can see its contents through the glistening membrane. Abdominal skin may cover the sac base and the umbilical cord is usually attached to its apex or slightly to the side. Large omphaloceles, measuring 5 cm or more in greatest dimension, may contain the stomach, liver, spleen, pancreas, and intestines. They may also contain segments of duplicated bowel. Smaller omphaloceles usually only contain intestines.
The function and histology of the displaced organs tends to be normal unless there is a coexisting congenital abnormality. The lining of the sac that covers the eviscerated organs consists of peritoneum internally and amnion externally.
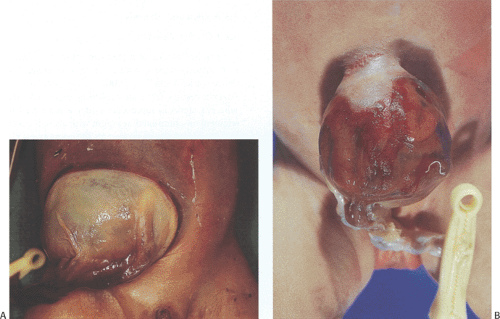 FIG. 6.46. Omphalocele. A: A large abdominal defect is covered by a thick white membrane extending into the base of the umbilical cord (dark red structure with clamp across it). The organs inside the omphalocele cannot be seen. B: The abdominal wall defect is smaller than that seen in A, and a clear sac covers the herniated intestines. A whitish membrane is forming near the abdominal wall attachment. At the periphery of this whitish lesion and within the clear membranous sac is an erythematous zone. The lesion continues into the umbilical cord, which has an umbilical clamp on it. |
Gastroschisis
Gastroschisis is the persistent herniation of abdominal viscera through an abdominal wall defect at the base of the umbilicus. The abdominal organs remain outside the abdominal cavity. No peritoneal sac or amniotic remnant covers the eviscerated abdominal contents. The incidence of gastroschisis has risen, increasing from 0.48 per 10,000 births in 1980 to 3.16 per 10,000 in 1993 (113,114). Young, socially disadvantaged women have the highest risk of having a child with gastroschisis (115). An increased risk of gastroschisis also associates with intrapartum use of recreational drugs or smoking (113), exposure to salicylates, and radiation (116). Gastroschisis predominates among male infants. It is often an isolated lesion (109). Sixteen to twenty percent of infants have associated congenital malformations, including total intestinal atresia (113,117,118).
Gastroschisis probably results from vascular injury and ischemia of the abdominal wall during the 5th to 11th fetal weeks, leading to defective somatopleural mesenchymal differentiation (118). The ischemia results from intrauterine disruption of the right omphalomesenteric artery. Exposure of the bowel to inflamed amniotic fluid (119) leads to perivisceritis and premature birth. The damaged bowel frequently develops secondary motility problems and malabsorption, which may present even following surgical repair. Other complications include obstruction due to coexisting malrotation, diverticulitis, ischemia, and perforation. Patients with ectopic gastric mucosa in the eviscerated bowel may develop a GI hemorrhage.
Gastroschisis may involve only the intestines or it may affect many other organs. Parts of the stomach, small intestine, and colon herniate through an abdominal wall defect, usually to the right of the umbilical cord (Fig. 6.47). All infants with gastroschisis have coexisting nonrotation and abnormal intestinal fixations. The small intestines typically appear thickened and shortened.
Omphalocele and gastroschisis are both associated with increased maternal serum and amniotic fluid α-fetoprotein (AFP) levels. Acetyl cholinesterase is also nearly always detectable in the amniotic fluid, albeit at much lower concentrations than in open neural tube defects (120). Antenatal ultrasound often allows an accurate diagnosis of gastroschisis.
The histology of the various organs may be normal (rarely) or changes may be present reflecting the presence of associated congenital abnormalities, heterotopias, atresia, meconium peritonitis, or perivisceritis. The last two entities lead to gastrointestinal wall thickening, serosal edema, and
fibrinous exudates and fibrosis. The intestinal muscularis propria may also become hypertrophic. Acute inflammation consisting predominantly of neutrophils and mononuclear cells may be present in gastroschisis and may lead to postnatal bowel dysfunction. Functionally, there is often malabsorption and hypomotility as the result of the inflammation.
fibrinous exudates and fibrosis. The intestinal muscularis propria may also become hypertrophic. Acute inflammation consisting predominantly of neutrophils and mononuclear cells may be present in gastroschisis and may lead to postnatal bowel dysfunction. Functionally, there is often malabsorption and hypomotility as the result of the inflammation.
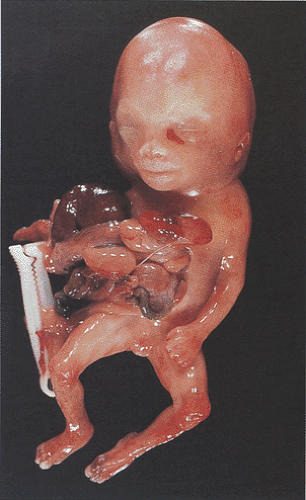 FIG. 6.47. Gastroschisis. This infant shows external herniation of most of the abdominal contents, including the liver, spleen, and intestinal tract. |
Long-term outcome in the absence of major chromosomal and structural abnormalities is excellent (121). Patient prognosis depends in part on whether the gastroschisis is an isolated lesion or whether there are associated abnormalities. The goal of the surgeon in both gastroschisis and omphalocele is to accomplish abdominal wall closure in a single stage. An alternate approach is to perform a staged closure using prosthetic materials while maintaining adequate nutritional support.
Bands
Peritoneal bands, known as Ladd bands, extend from the cecum, ascending colon, or posterior wall transduodenally to the subhepatic region, compressing the duodenum and causing partial obstruction, vascular compression, and intestinal ischemia. The bands represent incomplete absorption of the cecal and ascending colonic mesentery. Limb–body wall malformations, also known as amniotic band syndrome (Fig. 6.48), cause body wall, limb, and intestinal malformations.
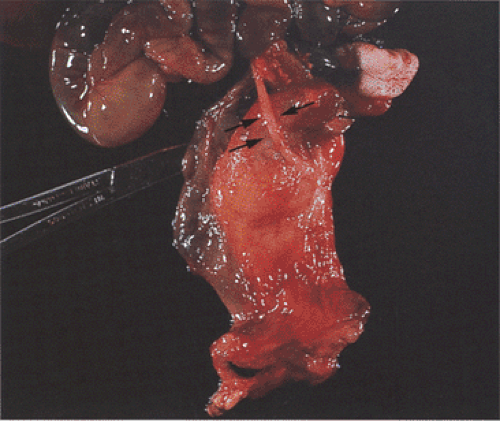 FIG. 6.48. Amniotic band syndrome. The fetus shows numerous abnormalities, including loss of limbs, an abdominal wall defect, gastroschisis, and an amniotic band (arrows). |
Atresia and Stenosis
Intestinal atresia and stenosis cause intestinal obstruction. Atresia occurs more often than stenosis. Their incidence varies from 1 per 2,000 to 6,000 live births (122). Atresia results from an occluding mucosal diaphragm, whereas stenosis represents either a narrowed intestinal segment or a luminal diaphragm with a small central opening. Duodenal atresia is the most common small intestinal atresia, followed by jejunal and ileal lesions. Duodenal atresia is less common than duodenal stenosis. Jejunoileal atresias affect 1 per 500 to 2,000 live births. Some patients have multiple atresias (123).
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


