CHAPTER 59 Chronic Pancreatitis
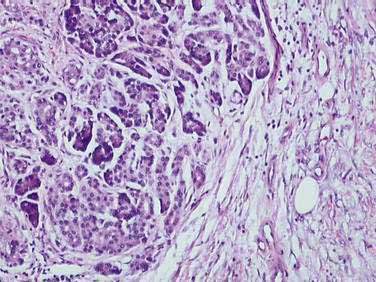
The traditional definition of chronic pancreatitis has been permanent and irreversible damage to the pancreas, with histologic evidence of chronic inflammation, fibrosis, and destruction of exocrine (acinar cell) and endocrine (islets of Langerhans) tissue (Fig. 59-1). As a consequence of this histologic damage, varying degrees of permanent and irreversible clinical, morphologic, and functional derangements occur. Chronic pancreatitis was distinguished from acute pancreatitis, characterized by complete recovery of the pancreas after the acute episode. This definition, using histologic criteria, is imperfect for a number of reasons. Most important, pancreatic tissue is rarely available to clinicians. Patients may have histologic evidence of chronic pancreatitis and have no symptoms or complications from the disease. In addition, the histologic features of chronic pancreatitis are often focal, such that a small biopsy, even if available, might miss the disease. Finally, the individual histologic features that are consistent with chronic pancreatitis are not unique, and may be seen in other conditions (such as normal aging). Alternatively, chronic pancreatitis has been defined based on clinical features (pain and exocrine or endocrine insufficiency) or on imaging techniques including ultrasonography (US), computed tomography (CT), endoscopic ultrasonography (EUS), magnetic resonance imaging (MRI), and endoscopic retrograde cholangiopancreatography (ERCP).
Finally, these staging systems make the assumption that acute pancreatitis and chronic pancreatitis are entirely separate entities, when in fact there is now abundant evidence documenting the evolution in many patients from acute to chronic pancreatitis (see Chapter 58). It is probably most accurate to think of the two conditions as separate ends of the same spectrum; acute pancreatitis is an identifiable event, whereas chronic pancreatitis is an ongoing process of variable tempo.
More recent reviews recognize these shortfalls and try to emphasize the importance of etiology, the difficulty of obtaining pancreatic tissue, and the lack of sensitivity of currently available diagnostic tools.1 These proposals have some advantages in that they attempt to define the disease in a more clinically useful manner, classify disease on the basis of etiology, and include advances in technology (e.g., EUS, magnetic resonance cholangiopancreatography [MRCP], genetic analysis) as well as functional and structural damage to the pancreas in the staging criteria.
EPIDEMIOLOGY
Chronic pancreatitis can be demonstrated in up to 5% of autopsies.2,3 Similar, though less pronounced, histologic features are seen even more commonly.4,5 Determining the prevalence of chronic pancreatitis from this type of autopsy data is misleading because the patients from whom the data are taken may not have had clinical symptoms of chronic pancreatitis during life. Long-standing alcohol use, even in moderate amounts, can lead to histologic changes of chronic pancreatitis without symptoms of chronic pancreatitis.6,7 Similarly, aging, chronic kidney disease, and long-standing diabetes mellitus can induce histologic changes within the pancreas that are difficult to distinguish from those of chronic pancreatitis.4,5 Making a diagnosis solely on the basis of autopsy data will therefore overestimate the rate of clinically important (i.e., symptomatic) chronic pancreatitis.
Estimates of annual incidence of chronic pancreatitis in several retrospective studies range from 3 to 9 cases per 100,000 population.8,9 The only prospective study, which was largely limited to alcoholic chronic pancreatitis, noted an annual incidence of 8.2 cases per 100,000 population and an overall prevalence of 27.4 cases per 100,000.10 A nationwide cross-sectional survey in Japan revealed an overall prevalence rate of 35.5 cases per 100,000 population with an estimated incidence of 14.4 per 100,000 and a male-to-female ratio of 3.5 : 1.11 A similar survey in France noted an annual estimated incidence of 7.7 per 100,000 population and a prevalence of 26 per 100,000.12 In most studies, alcohol abuse accounts for two thirds of all cases of chronic pancreatitis. These limited epidemiologic data demonstrate substantial geographic variation.13 The variation may partly be due to differences in alcohol consumption in different populations, but another part of the variation in incidence rates may merely reflect different diagnostic approaches and different diagnostic criteria.
Chronic pancreatitis accounts for substantial morbidity and health care costs. Approximately 26,000 hospital admissions to non-federal hospitals with a first-listed diagnosis of chronic pancreatitis occur yearly; in more than 80,000 yearly admissions, chronic pancreatitis is listed as one of the discharge diagnoses.14,15 The prognosis of chronic pancreatitis is variable and is driven largely by the presence of ongoing alcoholism in persons with chronic alcoholic pancreatitis and equally by concomitant tobacco use. One can estimate prognosis from such features as need for medical care or hospitalization or from the development of complications, reduced quality of life, or mortality.
Data on the quality of life of patients with chronic pancreatitis16–19 document that the presence of abdominal pain and alcohol abuse (in those with alcoholic chronic pancreatitis) are the dominant negative influences on quality of life and that, not surprisingly, quality of life is substantially worse for such patients than for the general population. Mortality in patients with chronic pancreatitis is also substantially influenced by the presence of continued alcoholism. In one large multicenter study, the standardized mortality ratio was 3.6:1 (i.e., those with a diagnosis of any form of chronic pancreatitis died at 3.6 times the rate of age-matched controls), and older subjects, those who smoked, and those with alcoholic chronic pancreatitis had the most significant reduction in survival.20 Continuing alcohol use raised mortality risk by an additional 60%. Overall, 10-year survival in patients with chronic pancreatitis is about 70%, and 20-year survival about 45%. Similar mortality data were noted in an analysis from Japan.21 The cause of death in patients with chronic pancreatitis usually is not the pancreatitis itself but other medical conditions commonly associated with smoking, continued alcohol abuse, pancreatic carcinoma, and postoperative complications.
PATHOLOGY
The different etiologies of chronic pancreatitis usually produce similar pathologic findings (see Fig. 59-1), particularly as the disease progresses. In early chronic pancreatitis the damage is varying and uneven. Areas of interlobular fibrosis are seen, with the fibrosis often extending to the ductal structures. Infiltration of the fibrotic area and lobules with lymphocytes, plasma cells, and macrophages is seen.22 The ducts may contain eosinophilic protein plugs. In affected lobules, acinar cells are surrounded and replaced by fibrosis. The islets are usually less severely damaged until very late in the course of the disease. Features of acute pancreatitis also may be seen, such as edema, acute inflammation, and acinar cell or fat necrosis. As the disease progresses, fibrosis within the lobules and between lobules becomes more widespread. The pancreatic ducts become more abnormal with progressive fibrosis, stricture formation, and dilation. The ductal protein plugs may calcify and obstruct major pancreatic ducts. Ductal epithelium may become cuboidal, may develop atrophy or squamous metaplasia, or may be replaced by fibrosis entirely. If special stains are performed, activated pancreatic stellate cells may be identified in close association with fibrosis.
These histologic features are found in most forms of chronic pancreatitis. Many of these changes, in particular perilobular fibrosis and ductal metaplasia, are also commonly seen in patients of advanced age without chronic pancreatitis, and in patients with long-standing diabetes mellitus.4,5 Obstructive chronic pancreatitis (associated with obstruction of the main pancreatic duct by a tumor or stricture) can differ slightly, in that the histologic changes are limited to the gland upstream of the obstruction and protein precipitates and intraductal stones are not seen.23
Autoimmune chronic pancreatitis demonstrates a more robust lymphoplasmacytic infiltrate, including plasma cells, and these are usually positive when stained for immunoglobulin G subtype 4 (IgG4).24 Obstructive phlebitis affecting the major and minor veins and a whorled fibrosis pattern are also characteristic, a pattern termed lymphoplasmacytic sclerosing pancreatitis (LPSP). A variant pattern termed idiopathic duct-centric chronic pancreatitis is characterized by additional neutrophilic infiltration. With time, the pattern may assume a more end-stage chronic pancreatitis appearance and become indistinguishable from other forms of chronic pancreatitis.
PATHOPHYSIOLOGY
The pathophysiology of chronic pancreatitis remains incompletely understood. The pathophysiologic processes must ultimately account for the features of chronic pancreatitis, including loss of parenchymal cells, self-sustaining chronic inflammation, and fibrosis. Any proposed mechanism must therefore include explanations for cellular necrosis or apoptosis, initiation and maintenance of inflammatory cell activation, and fibrogenesis by pancreatic stellate cells. The pancreas, like all other organs, has a limited repertoire of responses to injury and although it is not clear that all of the various etiologies have identical pathophysiology, the end histologic result is similar. The study of mechanisms of disease is hampered by the difficulty of obtaining tissue in humans and the relative lack of animal models of chronic pancreatitis,25,26 as opposed to acute pancreatitis.
Alcoholic chronic pancreatitis, being the most common form, has received the most attention. No single theory explains adequately why only about 10% of heavy alcohol users develop chronic pancreatitis. Alcohol is metabolized by the liver and the pancreas. In the liver the main end product of oxidative alcohol metabolism is acetaldehyde. In the pancreas, an alternative pathway produces fatty acid ethanol esters (FAEEs). Alcohol and its metabolites like FAEE, have direct injurious effects on pancreatic acinar cells. Increased membrane lipid peroxidation, a marker of oxidative stress and free radical production, can be seen in animal models and human alcoholic chronic pancreatitis.27–29 In addition FAEEs are able to induce sustained elevations in cytosolic calcium in acinar cells,29,30 a mechanism shared by other experimental causes of pancreatitis. Alcohol may also lead to pathologic increases in acinar cell sensitivity to physiologic stimuli such as cholecystokinin (CCK)31 and redirect CCK-mediated zymogen exocytosis to the basolateral rather than apical membrane.32 This basolateral secretion would place digestive enzymes in the interstitial space, where they could produce tissue damage. Chronic alcohol ingestion in animal models also alters expression of multiple genes in acinar cells, which could increase the sensitivity to physiologic stress33 and up-regulate the expression and activity of enzymes involved in cell death.34 Alcohol can promote the inflammatory responses involved in pancreatitis.35 These multiple effects of alcohol on the acinar cell are complemented by alcohol injury to ductal cells.36 Finally alcohol and its metabolites appear to stimulate the pancreatic stellate cell.36–38 These cells, as in the liver, appear to be the final common pathway for fibrosis.
Pancreatic stellate cells are found in association with the acinar units. They are typically found in the periacinar space, with long cytoplasmic processes extending to the acini themselves, but are also present in smaller numbers in association with blood vessels and ducts. Quiescent pancreatic stellate cells are recognized by the presence of vitamin A lipid droplets in the cytoplasm. When activated, they assume a stellate or myofibroblastic appearance, express smooth muscle actin, and lose the lipid droplets. This activation is necessary for the cell to begin to secrete extracellular matrix and produce fibrosis within the gland. Activation of pancreatic stellate cells can occur by alcohol or one of its metabolites, but also occurs in response to both inflammatory cytokines that are released following pancreatic acinar cell necrosis and to reactive oxygen species.36–38 In addition, growth factors (platelet-derived growth factor, transforming growth factor-β1), hormones, intracellular signaling molecules, transcription factors, and angiotensin II can activate pancreatic stellate cells.38 Activated pancreatic stellate cells are found in areas of extensive necrosis and inflammation in acute pancreatitis, in human as well as animal tissues. These activated pancreatic stellate cells produce autocrine factors that maintain the activated phenotype. In addition to their role in secretion and modulation of the extracellular matrix, pancreatic stellate cells can proliferate in response to stimulation, migrate to areas of inflammation, and participate in phagocytosis. Activation of pancreatic stellate cells is likely occurring via at least two mechanisms in alcoholic chronic pancreatitis: directly by alcohol and its metabolites and indirectly by cytokines induced by cellular necrosis.36
There have been several hypotheses for the pathophysiology of chronic pancreatitis that attempt to interweave these concepts into a coherent paradigm. One hypothesis focuses on the concept that ductal obstruction (from strictures or stones) is the cause rather than the effect of chronic pancreatitis. This hypothesis, the ductal obstruction hypothesis, is not consistent with most clinical and experimental evidence and with few exceptions (such as the rare condition of obstructive chronic pancreatitis) is not applicable to human chronic pancreatitis. A second paradigm, the toxic-metabolic hypothesis, focuses primarily on the role of alcohol and its metabolites (or other toxins) and their ability to damage the pancreas and activate pancreatic stellate cells. A third model that has been proposed is the necrosis-fibrosis hypothesis, which holds that the occurrence of repeated episodes of acute pancreatitis with cellular necrosis or apoptosis eventually leads to the development of chronic pancreatitis as the healing process replaces necrotic tissue with fibrosis. This last hypothesis has significant supporting evidence from some natural history studies that document the more common development of chronic pancreatitis in patients with more severe and more frequent acute attacks of alcoholic pancreatitis.39,40 The concept that multiple clinical or subclinical attacks of acute pancreatitis lead to chronic pancreatitis is certainly being reinforced by observations in both animal models and in humans.25,29,35,36,41
It is not clear why only a small subset of chronic alcoholics develop chronic pancreatitis. Some of the many possible explanations might include the presence of important co-toxins, differences in the genetic or epigenetic background, or differences in the microenvironment in the pancreas. For example, tobacco use is one very important cofactor for the development of alcoholic (and other types) chronic pancreatitis.42,43 There are also unexplained racial differences in the rates of alcoholic chronic pancreatitis.44 Multiple mutations have been identified in several forms of chronic pancreatitis, suggesting a complex genetic background that provides the relative predisposition to develop chronic pancreatitis (see Chapter 57). Mutations in the cystic fibrosis transmembrane conductance regulator (CFTR), cationic trypsinogen gene (PRSS1 gene), serine protease inhibitor Kazal type 1 (SPINK1, a trypsin inhibitor, formerly called PSTI or pancreatic secretory trypsin inhibitor), and many others have been identified.36,41,45–48 These mutations and the many more yet to be identified may be adequate in and of themselves to produce pancreatitis (at this point only PRSS1 satisfies that requirement) or may only predispose to disease. In the case of PRSS1, a number of families with hereditary pancreatitis (see Chapter 57) have been identified with mutations in the PRSS1 gene. These mutations appear to lead to a gain of function mutation in which trypsinogen, once activated to trypsin, is resistant to inactivation. The mutant trypsin might therefore be able to activate other proenzymes and to produce clinical or subclinical episodes of acute pancreatitis, ultimately leading to chronic pancreatitis in affected patients. This presumed pathophysiology provides support for the necrosis-fibrosis theory of chronic pancreatitis, in which repeated episodes of acute pancreatitis ultimately lead to chronic pancreatitis. Cystic fibrosis is associated with abnormalities of bicarbonate secretion by ductal cells and the eventual development of dilated pancreatic ducts, intraductal precipitates, and pancreatic atrophy (see Chapter 57). Several studies have documented an increase in rate of CFTR mutations in patients with presumed “idiopathic” pancreatitis.49–52 SPINK1 mutations have been found in idiopathic chronic pancreatitis, hereditary pancreatitis, tropical pancreatitis, and alcoholic pancreatitis.45,46,53–55 These mutations are discussed in detail in Chapter 57 but are useful to consider here as a basis for an evolving paradigm of pathogenesis This paradigm has been labeled SAPE—sentinal acute pancreatitis event—and conceptualizes a complex genetic background that provides the predisposition to disease.1,29,36,37,41,46,47 This background may include mutations in genes that code for digestive enzymes, protease-enzyme inhibitors, ion channels, and a variety of others including genes that affect the metabolism of environmental toxins (e.g., tobacco or alcohol), genes that have a role in inflammation or fibrosis, and others yet to be discovered. Although a few mutations may be sufficient to produce pancreatic damage in most or all who carry it, the majority of mutations identified to date are not that dominant. On this complex polygenic background is overlaid some environmental insult, such as chronic alcohol use, that applies physiologic stress to the acinar, ductal, and stellate cells. This stress may be insufficient to produce injury or may produce cellular necrosis or apoptosis. The initiating event for necrosis is the activation of digestive enzymes within the acinar cell, either by the toxic effect of the environmental insult or because of some underlying mutation that leads to excessive activation of trypsin (see Fig. 57-3). Inflammation follows the necrosis, and this necroinflammatory process may either progress or resolve. In some individuals, the situation may never progress beyond this stage. In others, continued cell metabolic and oxidative stresses or some other trigger could produce continuing or repeated acinar cell injury with necrosis. This process, as is believed to be the case in other organs, could be associated with the activation of stellate cells. The acute inflammatory response could resolve, or, alternatively, the inflammatory response might switch to an anti-inflammatory response, with resident macrophages producing cytokines and activated stellate cells laying down extracellular matrix, with the formation of fibrosis. Fibrosis would start a vicious circle by causing additional acinar cell ischemia and continuing to drive the inflammatory–anti-inflammatory cycle. This type of hypothesis could theoretically explain many forms of chronic pancreatitis. This framework seems to fit all the developing experimental and clinical data and is a useful way in which to think about the pathophysiology of chronic pancreatitis: as a disease associated with a variety of different genetic predispositions, a variety of disease triggers, and a similar final common pathway producing pancreatic injury and fibrosis, ultimately with organ failure.
ETIOLOGY (Table 59-1)
ALCOHOL
In Western countries, alcohol is the cause of at least 70% of all cases of chronic pancreatitis.8–10,14,56–60 The risk of alcoholic chronic pancreatitis increases logarithmically with rising alcohol use, but there is no true threshold value below which the disease does not occur.48,60–63 Therefore, in countries with widespread alcohol consumption, it may be difficult to determine with certainty whether alcohol contributed to or caused the disease. In nearly all patients, at least 5 years (and in most patients more than 10 years) of intake exceeding 150 g/day (a standard drink contains about 14 g of alcohol) are required before the development of chronic pancreatitis. Only 3% to 15% of heavy drinkers ultimately develop chronic pancreatitis, suggesting an important cofactor.56,61,62,64 Potential cofactors include genetic polymorphisms and mutations,27–29,47,48,64,65 a diet high in fat and protein,47,61,64 the type of alcohol or manner of ingestion,62 an associated relative deficiency of antioxidants or trace elements,66,67 and smoking.42,47,56,61,64,68,69 Of these, smoking appears to be the strongest association. In some studies, 90% of those who develop alcoholic chronic pancreatitis are also chronic smokers.64,68,69 Smoking also appears to predispose to more rapid development of pancreatic calcification.42,43,62,70,71 There are also racial differences in the risk for development of alcoholic pancreatitis, perhaps suggesting some difference in the ability to detoxify environmental toxins or alcohol or other genetic or epigenetic factors.44,47,62,64 Although the risk of alcoholic chronic pancreatitis is higher in blacks,44 data from self-reported surveys of alcohol use demonstrate that the proportion of blacks who drink alcohol or smoke is actually lower than that in whites.72
Table 59-1 Classification of Chronic Pancreatitis
CFTR, cystic fibrosis transmembrane conductance regulator gene; PRSS1, protease serine 1 gene; SPINK1, serine protease inhibitor, Kazal type 1 gene.
Many patients with alcoholic chronic pancreatitis have an early phase of recurrent attacks of acute pancreatitis, which may last five or six years, followed by the later development of chronic pain or exocrine or endocrine insufficiency. It has been believed, based on several large natural history studies, that most patients who present with their first attack of acute alcoholic pancreatitis have already developed histologic chronic pancreatitis. Up to 40% of patients presenting with acute alcoholic pancreatitis, however, do not progress to clinically identifiable chronic pancreatitis (calcification and exocrine or endocrine insufficiency) even with very long follow-up, and may not even have recurrent attacks.39,62,64,73–75 Based on autopsy studies6,7 and functional studies,76 however, it is likely these patients actually have histologic evidence of chronic pancreatitis, although they may not have developed a recognizable chronic clinical illness.
Not all patients with alcoholic chronic pancreatitis present with acute episodes of pancreatitis. Less than 10% of patients present with exocrine or endocrine insufficiency in the absence of abdominal pain.57–5964 Some present with chronic pain in the absence of antecedent acute attacks of pain. Cessation of alcohol use after the onset of alcoholic pancreatitis appears to diminish the rate of progression to exocrine insufficiency and endocrine insufficiency but does not halt it.77 Stopping alcohol does seem to reduce the chance of recurrent attacks of acute alcoholic pancreatitis in those who have not yet developed obvious chronic pancreatitis.75
The prognosis of alcoholic chronic pancreatitis is relatively poor,20,21 and mortality is generally greater than that seen in chronic pancreatitis of other etiologies. Quality of life is also substantially diminished. Pain generally continues for years, although it may spontaneously remit. Exocrine and/or endocrine insufficiency develops in many patients, although this process may take several years. In one large natural history study, exocrine insufficiency developed in 48% of patients at a median of 13.1 years after presentation, whereas endocrine insufficiency developed in 38% after a median of 19.8 years after presentation.58 Diffuse pancreatic calcifications developed in 59% at a median of 8.7 years after diagnosis. Other studies have noted more rapid and more frequent development of calcifications, exocrine insufficiency, and endocrine insufficiency.57
TOBACCO
Exposure to tobacco smoke can induce pancreatic damage in animals.78 As mentioned, smoking is common in patients with alcoholic chronic pancreatitis and is associated with an increased risk for pancreatic calcifications, and smoking cessation after the clinical onset of chronic pancreatitis reduces the risk of subsequent calcifications.79 There is also evidence that smoking is an independent risk factor for chronic pancreatitis.80,81 Smoking is also associated with a much higher rate of secondary pancreatic cancer and overall mortality in patients with chronic pancreatitis.11,20
TROPICAL PANCREATITIS
Tropical pancreatitis is one of the most common forms of chronic pancreatitis in certain areas of southwest India. It has been reported from a number of other areas, including Africa, southeast Asia, and Brazil. The disease as initially described is essentially restricted to areas within 30 degrees of latitude from the equator. Tropical pancreatitis is generally a disease of youth and early adulthood, with a mean age at onset of 24 years.82–84 More than 90% of patients have the illness before 40 years. The overall prevalence from surveys in an endemic area (southern India) is 1 in 500 to 1 in 800 population.82,85 Tropical pancreatitis accounts for about 70% of all cases of chronic pancreatitis in southern India, whereas alcohol is a more dominant cause in the north. Surveys in Kerala province in southwest India have noted that there is a shift toward older age at presentation, less malnutrition, and less severe diabetes.86 These later surveys also note higher rates of alcohol abuse in this area and alcohol gradually becoming a more common cause of chronic pancreatitis.
The pathophysiology of tropical pancreatitis is unknown. As discussed in Chapter 57, a number of genetic mutations have been identified, with mutations in the SPINK1 and chymotrypsinogen genes being most common.87 Environmental triggers for the disease that have been proposed include protein-calorie malnutrition, deficiencies of trace elements and micronutrients coupled with oxidative stress (via xenobiotics, industrial pollutants, diet, or nutritional deficiency), cyanogenic glycosides present in cassava (tapioca—a main dietary component), viral and parasitic infections, and others.
GENETIC
Only one type of mutation appears sufficient to cause chronic pancreatitis: mutations in PRSS1 in families with hereditary pancreatitis. All other identified mutations (discussed in Chapter 57) and polymorphisms should be considered as cofactors, mutations that predispose to disease by increasing susceptibility, or as modifier genes, that increase the pace or severity of disease. It is certainly possible that mixtures of polymorphisms and mutations work together to determine the susceptibility to disease. The most commonly identified mutations occur in the PRSS1 (cationic trypsinogen), SPINK1 (trypsin inhibitor), and CFTR genes. Several studies have suggested that certain less severe CFTR gene mutations and SPINK1 mutations may be associated with idiopathic chronic pancreatitis. CFTR gene mutations have been identified in up to half of patients with idiopathic chronic pancreatitis.46,49–52 This proportion is far greater than expected within this population. Analysis of these data has suggested that the combination of a more severe CFTR mutation on one chromosome with a mild mutation on the other is particularly associated with chronic pancreatitis (see Chapter 57). These studies examined only the most common CFTR mutations. There are now more than 1200 known CFTR mutations, and further studies are needed to define their contribution to idiopathic and other forms of chronic pancreatitis. SPINK1 mutations have also been identified with increased frequency in patients with unexplained chronic pancreatitis, including some mixed heterozygotes with combined CFTR and SPINK1 mutations. Genetic testing for these mutations is commercially available.
AUTOIMMUNE PANCREATITIS
Autoimmune pancreatitis refers to a distinct chronic inflammatory and sclerosing disease of the pancreas. The distinct characteristic of the disease is a dense infiltration of the pancreas, and often other organs, with lymphocytes and plasma cells (Fig. 59-2A), many of which express IgG4 on their surface. The autoimmune target of this IgG4 and the trigger for disease are unknown. However, recently a protein expressed in pancreatic acinar cells, UBR2 (ubiquitin-protein ligase E3 component n-recognin 2) has been proposed to be the target of the antibody.87a Of interest, the antibodies also react to a protein of Helicobacter pylori, PBP (plasminogen-binding protein), suggesting a role for H. pylori infection in autoimmune pancreatitis.
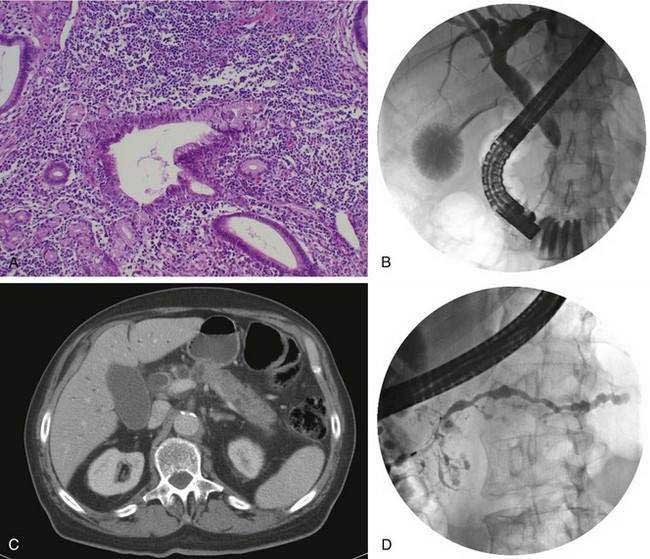
Fibrosis, sclerosis, and obliterative phlebitis are characteristically seen in association with the chronic inflammatory infiltrate. Although this inflammatory infiltrate is present in the pancreas, similar infiltrates may be seen in the bile duct, salivary glands, retroperitoneum, lymph nodes, kidney, prostate, ampulla, and occasionally other organs.24 An equally characteristic feature of this disease is the often rapid response to glucocorticoid therapy. Much of the information on this disease comes from a series of studies from Japan and other Asian countries. Early reports noted chronic pancreatitis characterized by the presence of autoantibodies, elevated levels of serum immunoglobulins, enlargement of the pancreas (diffuse or focal), pancreatic duct strictures, and pathologic features of a dense lymphocytic infiltrate.88
Studies from Japan note that up to 6% of all patients evaluated for chronic pancreatitis have autoimmune pancreatitis, and the overall prevalence is estimated to be 0.82 per 100,000 persons.24,88–92 The disease may occur in an isolated form or may be associated with extrapancreatic manifestations. The most common extrapancreatic conditions identified include biliary strictures, hilar lymphadenopathy, sclerosing sialadenitis, retroperitoneal fibrosis, and tubulointerstitial nephritis.24,88–93 Autoimmune pancreatitis may therefore be one manifestation of what has been termed IgG4-related sclerosing disease or IgG4-related systemic disease. There also appears to be some overlap with an unusual variant of Sjögren’s disease termed Mikulicz’s disease, in which a massive IgG4-positive mononuclear infiltrate is seen in the salivary and lacrimal glands.94
The disease is seen more commonly in men (2 : 1) and usually manifests in middle age. More than 85% of patients present after the age of 50 years. One of the most common presentations is painless obstructive jaundice due to obstruction of the intrapancreatic bile duct (see Fig. 59-2B). Jaundice may occur from compression of the bile duct by the enlarged pancreas or by infiltration of the biliary tree by the chronic inflammatory process. Additional symptoms may include weight loss, vomiting, and glucose intolerance.24 Although pain is not frequently present, abdominal and referred back pain may occur. These clinical features, coupled with imaging studies demonstrating diffuse or focal pancreatic enlargement (see Fig. 59-2C), often raise the suspicion of pancreatic adenocarcinoma. In studies in the United States in patients who underwent pancreatic resection for presumed pancreatic carcinoma but were found to have no malignancy in the resected specimen, up to 11% show evidence of autoimmune pancreatitis.95 Jaundice or cholestasis may also occur due to strictures of the biliary tree in other locations. A pattern similar to that seen in primary sclerosing cholangitis (PSC) is seen, with a predilection for involvement of the hilar region. The pattern may mimic not only PSC but also cholangiocarcinoma. The disease, unlike classic PSC, is not associated with inflammatory bowel disease and is steroid-responsive (see Chapter 68). Additional clinical manifestations include a sclerosing sialadenitis (usually presenting as bilateral symmetrical swelling of the salivary glands), retroperitoneal fibrosis (most commonly presenting as hydronephrosis due to entrapment of the ureters), tubulointerstitial nephritis, lymphadenopathy (particularly mediastinal, cervical, and abdominal), prostatitis, and sclerosing cholecystitis, interstitial pneumonia, pseudotumors of the liver, lung, and pituitary.
Abdominal ultrasonography usually shows a diffusely enlarged and hypoechoic pancreas. The appearance on EUS is similar. CT most commonly reveals a diffusely enlarged sausage-shaped pancreas (see Fig. 59-2C) in which enhancement with the intravenous contrast agent is delayed and prolonged.24,91,96 Some patients may have a capsule-like low-density rim around the pancreas in delayed images. Focal swelling can also occur, mimicking a pancreatic mass.24,






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


