CHAPTER 3 Cellular Growth and Neoplasia
MECHANISMS OF NORMAL CELL HOMEOSTASIS
CELLULAR PROLIFERATION
Neoplasia is the ultimate result of the disruption of exquisite mechanisms regulating normal cell growth. Growth is determined by the balance of cellular proliferation, differentiation, senescence, and programmed cell death. Proliferation occurs as cells traverse the cell cycle (Fig. 3-1). In preparation for cell division, there is a period of deoxyribonucleic acid (DNA) synthesis, designated the S phase. After an intervening gap period, designated the G2 phase, actual mitosis occurs in the M phase. After another intervening gap period, the G1 phase, DNA replication can begin again.
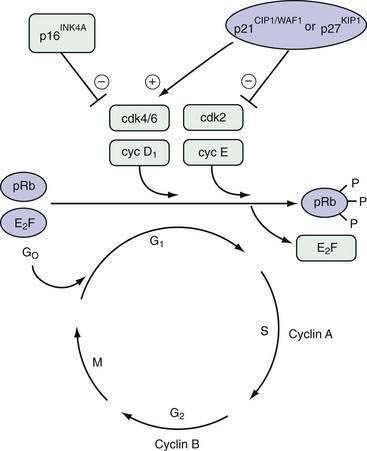
The commitment to proceed through DNA replication and cell division occurs during the G1 phase at the so-called start or restriction (R) point. Cells may exit this cycle of active proliferation before reaching the R point and enter a quiescent phase, G0. Cells can subsequently re-enter the cell cycle from the G0 state (see Fig. 3-1). The duration of each cell cycle phase as well as the overall length of the cycle vary among cell types.
Regulation of cell cycle progression appears to be achieved principally by cyclins and cyclin-dependent kinase activity at the G1/S and G2/M phase transitions. Cyclin proteins are classified on the basis of their structural features and temporal expression patterns during the cell cycle (see Fig. 3-1). Cyclins A and B are expressed predominantly during the S and G2 phases. In contrast, cyclins D and E proteins are most active during the G1 phase.1 Overexpression of cyclin D1 in fibroblasts results in more rapid entry of cells into the S phase. Cyclin D1 is frequently overexpressed in a number of GI and non-GI malignancies, including those originating from the oral cavity, esophagus, breast, and bladder.2
Each cyclin forms a complex with a cyclin-dependent kinase (cdk) in a cell cycle–dependent fashion. Cyclins function as catalysts for cdk activity (see Fig. 3-1). Cdks physically associate with cyclins through their catalytic domains. The cyclin-cdk complexes regulate cell cycle progression through phosphorylation of key target proteins, including the retinoblastoma gene product (pRb) as well as the Rb family members p130 and p107.3 The final result is progression out of G1 into the S phase of the cell cycle.
The cell cycle is also regulated by multiple cdk inhibitors; p21CIP1/WAF1 and p27KIP1 are inhibitors of cyclin E/cdk2. Originally discovered to be part of the complex containing cyclin D1 and cdk4/6, p21CIP1/WAF1 is transcriptionally activated by the TP53 tumor suppressor gene product (see Fig. 3-1).4 p16INK4A is another cdk inhibitor that specifically inhibits cdk4 and cdk65 and is part of a larger family of related inhibitors that includes p14, p15, and p18. p16INK4A is frequently inactivated in esophageal squamous cell cancers and pancreatic ductal adenocarcinomas, a finding that is consistent with its function as a tumor suppressor gene.6,7 p16INK4A disrupts the complex of cyclin D1 and cdk 4/6, thereby freeing p21CIP1/WAF1 and p27KIP1 to inhibit the activity of cyclin E/cdk2.8
PROGRAMMED CELL DEATH AND SENESCENCE
Studies of the roundworm Caenorhabditis elegans have led to the initial identification of the gene ced-3, a protease that is the major effector of apoptosis. Two key regulators of ced-3, designated ced-9 and ced-4, were found to prevent or induce apoptosis, respectively.9 The mammalian oncogene bcl-2 shares homology with ced-9 and protects lymphocytes and neurons from apoptosis10; bcl-2 complexes with bax, a protein that by itself contributes to apoptosis.11 Of note, both bcl-2 and bax are part of larger gene families, and the stoichiometric relationships among different combinations of the encoded proteins can determine the balance between cell survival and cell death.12
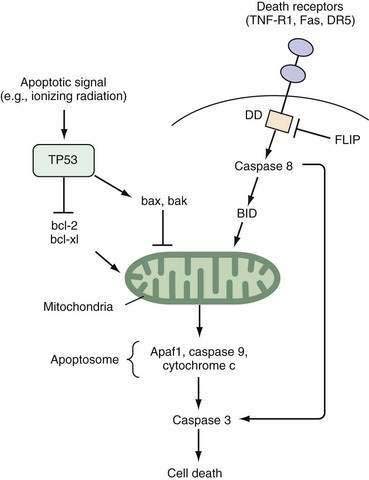
Two well-defined pathways that trigger apoptosis have been described in detail. One pathway is mediated through membrane-bound death receptors, which include tumor necrosis factor (TNF) receptors, Fas, and DR5, whereas the other pathway involves activation of TP53 expression by environmental insults such as ionizing radiation, hypoxia, or growth factor withdrawal, with a subsequent increase in the bax-to-bcl-2 ratio. Both pathways converge to disrupt mitochondrial integrity and release of cytochrome c (Fig. 3-2). The so-called apoptosome complex (cytochrome c, caspase 9, and Apaf1) then activates downstream caspases, such as caspase 3, eventuating in cell death. Activation of caspases, intracellular cysteine proteases that cleave their substrates at aspartate residues, is a key step in programmed cell death in mammalian cells.
Replicative senescence also plays a role in determining overall growth in cell populations. Most primary cells when grown in vitro have a limited replicative potential and eventually undergo senescence.13 In contrast, malignant cells can replicate indefinitely. Up-regulation of the telomerase enzyme is essential to escape from replicative senescence. Telomeres are repetitive DNA sequences at the ends of all chromosomes that regulate chromosomal stability. Telomeres shorten with each cell division and, when they have been reduced to a certain critical length, senescence occurs. Cancer cells are able to maintain their telomere length despite multiple cell divisions through the reactivation of telomerase enzyme activity.14
SIGNALING PATHWAYS THAT REGULATE CELLULAR GROWTH
Cellular proliferation is achieved through transition of cells from G0 arrest into the active cell cycle (see Fig. 3-1). Although progression through the cell cycle is controlled by the regulatory proteins just described, overall proliferation is modulated by external stimuli. Growth factors that bind to specific transmembrane receptors on the cell surface may be especially important. The cytoplasmic tails of these transmembrane receptor proteins produce an intracellular signal after ligand binding.
Other receptors on the cell surface possess kinase activity directed toward serine or threonine residues rather than tyrosine. These receptors also phosphorylate a variety of cellular proteins, leading to a cascade of biological responses. Multiple sites of serine and threonine phosphorylation are present on many growth factor receptors, including the tyrosine kinase receptors, suggesting the existence of significant interactions among various receptors present on a single cell.15 The transforming growth factor-β (TGF-β) receptor complex is one important example of a serine-threonine kinase–containing transmembrane receptor.
Many receptors are members of the so-called seven-membrane–spanning receptor family. These receptors are coupled to guanine nucleotide binding proteins, and are designated G proteins. G proteins undergo a conformational change that is dependent on the presence of guanosine phosphates.16 Activation of G proteins can trigger a variety of intracellular signals, including stimulation of phospholipase C and the generation of phosphoinositides (most importantly, inositol 1,4,5-triphosphate) and diacylglycerol through hydrolysis of membrane phospholipids, as well as modulation of the second messengers cyclic AMP and GMP.17 Somatostatin receptors exemplify a G protein–coupled receptor prevalent in the GI tract.
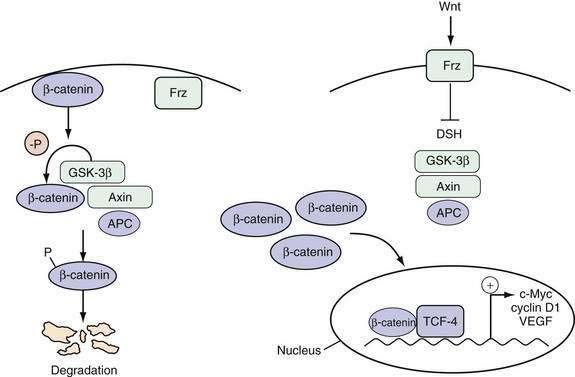
The Wnt pathway is one important example of a signaling pathway that regulates the cell cycle machinery to control the proliferation of intestinal epithelial cells (Fig. 3-3). Although the details of the specific interactions between the Wnt ligand and its receptor Frz, a member of the seven-membrane receptor family, in the GI tract are not fully clarified, an active Wnt signal ultimately results in the accumulation of β-catenin in the nucleus, where it binds with the transcription factor TCF-4 to activate a set of target genes.18 Inhibition of the Wnt signal in mice can be achieved by deletion of TCF-4 or overexpression of a Wnt inhibitor designated Dickkopf1, which results in dramatic hypoproliferation of the intestinal epithelium.19,20 This hypoproliferation appears to be mediated by decreased expression of the TCF-4 target gene c-MYC, which directly represses p21CIP1/WAF1.21 Thus, a Wnt signal stimulates proliferation of intestinal epithelial cells by repressing the cell cycle inhibitor p21CIP1/WAF1.
Cyclin D1 has an extremely short half-life (<20 minutes) and is a rate-limiting factor for progression through the G1 phase of the cell cycle (see Fig. 3-1). Consequently, it is one of the most tightly regulated of all cell cycle proteins. Extracellular signals from growth factors, including EGF, colony-stimulating factor 1 (CSF-1), platelet-derived growth factor (PDGF), and insulin-like growth factor (IGF), can regulate cellular proliferation by rapidly inducing the expression of the cyclin D1 gene.22
Tissue homeostasis is also maintained by growth-inhibiting signals that counterbalance proliferative signals. TGF-β is a potent growth-inhibiting factor that mediates arrest of the cell cycle at the G1 phase. TGF-β not only induces the transcription of the cell cycle inhibitors p15INK4B and p21CIP1/WAF1, but also enhances the inhibitory activity of p27KIP1 on the cyclin E/cdk2 complex (see Fig. 3-1).23 These effects are mediated intracellularly through the Smad family of proteins.
INTESTINAL TUMOR DEVELOPMENT: MULTISTEP FORMATION AND CLONAL EXPANSION
Multiple sequential genetic alterations are required for the transformation of normal intestinal epithelium to frank malignancy. This multistep nature of tumorigenesis is most directly illustrated by the changes that accrue in the development of colonic neoplasia (see Chapters 122 and 123). The accumulation of genetic alterations roughly parallels the progression from normal epithelium through adenomatous polyps (or, in the case of ulcerative colitis, flat dysplastic mucosa) to malignant neoplasia. Studies on the molecular pathogenesis of colon cancer have served as a paradigm for the elucidation of genetic alterations in other GI cancers. For example, a similar progression is also seen in the transition from normal squamous epithelium to metaplastic mucosa (Barrett’s esophagus) through dysplasia to adenocarcinoma of the esophagus. Gastric and pancreatic oncogenesis are each thought to proceed through similar multistep pathways.
Models of the multistep or multiple hit process of tumor formation have largely superseded earlier concepts of oncogenesis that discriminated between tumor initiation and subsequent promotion. Initiation was attributed to a single change in a cell that converted it from a normal to a malignant cell. Promotion reflected all the factors that acted after the initiating event to enhance tumor growth. However, oncogenesis occurs through a series of events that result in incremental changes in cell behavior until the cell eventually passes some threshold associated with the malignant phenotype. Nevertheless, there is still some merit in a more limited concept of tumor promotion. A number of factors promote the likelihood of malignant transformation through the stimulation of increased cellular turnover, which increases opportunities for somatic mutations to occur.24 In the GI tract, these promoting factors include dietary constituents (see later) as well as chronic inflammation, which are associated with increased cell proliferation. Thus, a number of chronic inflammatory conditions increase the site-specific risk of cancer, such as ulcerative colitis (see Chapter 112), chronic gastritis (see Chapters 51 and 54), chronic pancreatitis (see Chapters 59 and 60), Barrett’s esophagus (see Chapters 44 and 46), and chronic hepatitis (see Chapter 94). Although the mechanisms whereby inflammatory processes elicit eventual tumor development are incompletely understood, cytokines produced by inflammatory cells can stimulate tumor cells, leading to activation of nuclear factor-κB (NF-κB) in the tumor cells that can serve to inhibit apoptosis and stimulate proliferation.25
Clonal expansion is also essential to tumor development.26 Whereas germline mutations may lead to altered expression of a gene in all cells in a tissue, subsequent additional somatic mutations generally occur only in a small, largely random subpopulation of cells. Clonal expansion occurs if a specific gene mutation results in a survival advantage for the cell. A second round of clonal expansion occurs when a cell within this population sustains still another genetic alteration, which further enhances its growth properties. After several iterations, a final genetic alteration eventually confers a property that, together with the preceding genetic alterations, makes a cell malignant.
Recent evidence has led to the suggestion that cancer stem cells in tumors may play a central role in tumorigenesis. These cells are defined by the capacity for self-renewal and the ability to generate progeny that lack this capacity but manifest the characteristics of the heterogeneous lineages that comprise the tumor.27 Failure to eradicate the cancer stem cell population is posited to underlie tumor recurrences after chemotherapy. Precise identification of the cancer stem cell compartment has been possible for hematologic malignancies. However, comparable definitive proof of their presence in solid tumors, such as intestinal cancers, remains a challenge.28
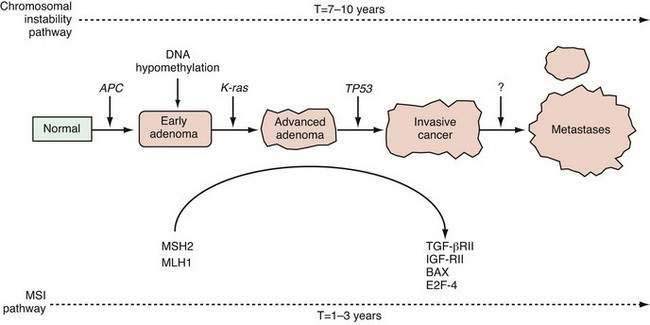
A genetically unstable environment was thought to be necessary for the development of the multiple alterations that ultimately result in cancer. Genomic instability is observed in almost all cancers, regardless of organ site. Instability of the genome may result from several mechanisms. In colon cancer, there are at least two well-recognized forms of genetic instability, and they have been termed chromosomal instability and microsatellite instability.29 Chromosomal instability results in tumor cells that display frequent aneuploidy, large chromosomal deletions, and chromosomal duplications. In contrast, tumors that display microsatellite instability are often diploid or near-diploid on a chromosomal level but harbor frequent alterations in smaller tracts of microsatellite DNA (see later discussion on DNA repair). Thus, there are at least two distinct routes to the formation of a colorectal cancer, depending on the nature of the underlying genetic instability (Fig. 3-4).
NEOPLASIA-ASSOCIATED GENES
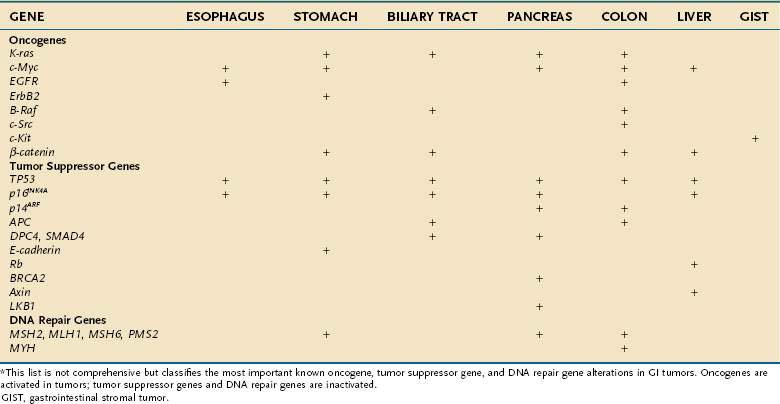
The genes that collectively play an important role in oncogenesis generally lead to disruption of the orderly mechanisms of normal cell proliferation. Insofar as normal cell proliferation appears to depend on a wide variety of genes, it is not surprising that alterations in diverse genes confer part or all the phenotypic features of transformation. Despite this diversity, all these genes that become altered appear to belong to one of three distinct groups: (1) oncogenes, which actively confer a growth-promoting property; (2) tumor suppressor genes, the products of which normally restrain growth or proliferation; and (3) DNA repair genes, which contribute to transformation by fostering genomic instabilbity and facilitating mutations in other genes. Activation of oncogenes or inactivation of tumor suppressor genes and DNA repair genes contributes to malignant transformation (Table 3-1).
Table 3-1 Oncogenes, Tumor Suppressor Genes, and DNA Repair Genes Altered in Gastrointestinal Tumors*
ONCOGENES
Oncogenes and Peptide Growth Factors
The transforming effects of enhanced expression of a variety of growth factors have been demonstrated both in vitro and in vivo. Several growth factor–related proteins encoded by oncogenes have now been recognized, including the family of Wnt proteins and Sis, which encodes the β chain of platelet-derived growth factor. It is axiomatic that cells that produce high levels of a growth factor must also express specific receptors activated by that growth factor to yield an autocrine growth-stimulating loop that is often present in neoplasms. For example, several colon-derived cancer cell lines express TGF-β and IGF I and II, as well as their receptors. This autocrine mechanism may be contrasted with overproduction of a growth factor that exerts its influence at a remote cellular target rather than within the tumor itself, exemplified by gastrin produced by gastrinoma cells, which exerts trophic effects on the gastric mucosa but not on the tumor itself (see Chapter 32).
Protein Kinase–Related Oncogenes
A brief consideration of receptor protein tyrosine kinase HER2/Neu/ERBB2, which is related to the EGF receptor, is particularly illustrative. There are four EGF receptor family members (ERBB1-4). The viral v-erb-b2 encodes a truncated form of the EGF receptor that lacks most of the external EGF-binding domain.30 As a result, the receptor no longer requires the presence of the ligand for activation and remains continuously activated, stimulating proliferation. The neu oncogene is derived from a rat cellular proto-oncogene closely related to the EGF receptor. The oncogene differs from its normal counterpart by a point mutation that changes a single residue (valine to glutamic acid) within the transmembrane domain, thereby causing activation of the 185-kd tyrosine kinase protein (p185neu).31 The human counterpart (ERBB2) of the neu oncogene is not mutated but is overexpressed or amplified in a variety of adenocarcinomas, including those arising in the stomach, breast, and prostate.32 In addition, ERBB2 expression increases progressively in the transition from normal esophageal mucosa through the dysplastic state characteristic of Barrett’s esophagus to esophageal adenocarcinoma.33
In contrast with the receptor type of tyrosine kinase that possesses intrinsic catalytic activity, many other receptors and membrane proteins lack self-contained signaling activity. Instead, they are coupled to nonreceptor tyrosine kinases on the cytoplasmic side of the plasma membrane that act as signal transducers. A number of oncogenes associated with neoplasms of the GI tract, most notably the colon, are members of the src family of nonreceptor tyrosine kinases. Members of the src family are approximately 60-kd phosphoproteins (v-src) that possess inherent tyrosine kinase activity and associate with the inner surface of the plasma membrane. Autophosphorylation of the normal c-src leads to attenuation of its kinase activity, thereby providing inherent regulation to limit unrestrained activity.34 Increased levels of c-src activity have been found in colonic cancer tissue and colon cancer–derived cell lines.35 Activating mutations of c-src have been identified in a subset of advanced, metastatic colon cancers.36









