Calcium, Phosphorus, and Vitamin D in Kidney Disease
Ishir Bhan
Ravi Thadhani
CHANGES IN CALCIUM AND PHOSPHOROUS METABOLISM IN CHRONIC KIDNEY DISEASE
A host of metabolic changes occur as chronic kidney disease (CKD) progresses, particularly with respect to mineral metabolism. As glomerular filtration rate (GFR) declines it is accompanied by a fall in the activity of the renal vitamin D 1α-hydroxylase enzyme. As a result, conversion of vitamin D from its inactive form (25-hydroxyvitamin D) to the active 1,25-dihydroxyvitamin D is impaired. With reduced 1,25-dihydroxyvitamin D activity, the vitamin D-dependent calcium absorption from the gastrointestinal tract is limited and blood concentrations of calcium can fall. Phosphorous excretion, in contrast, becomes restricted in the setting of CKD because of decreased tubular function. Reduced phosphorous excretion appears also to trigger increased production of fibroblast growth factor-23 (FGF-23). This recently discovered phosphaturic hormone also suppresses 1α-hydroxylase, limiting the production of 1,25-dihydroxyvitamin D.
In addition to the effects of declining kidney function and FGF-23 on production of 1,25-dihydroxyvitamin D, its synthesis may also be limited by a reduction in substrate for 1α-hydroxylase. Indeed, deficiency of 25-hydroxyvitamin D is common in CKD. This may result at least in part from the urinary loss of the vitamin D binding protein (DBP) and 25-hydroxyvitamin D bound to DBP in individuals with proteinuria. In addition, the ability of the skin to produce vitamin D in response to ultraviolet radiation appears to be impaired in CKD. Protein malnutrition in this population may also play a role in promoting 25-hydroxyvitamin D deficiency.
One of the roles of 1,25-dihydroxyvitamin D is to suppress the production of parathyroid hormone (PTH) gene transcription. Not only is 1,25-dihydroxyvitamin D synthesis impaired in CKD, but the amount of vitamin D receptor (VDR) in the parathyroid gland is also reduced. Furthermore, in advanced CKD the binding of 1,25-dihydroxyvitamin D to VDR and of VDR to vitamin D response elements (VDRE) in DNA are also decreased. As a result of decreased vitamin D action, including reduced serum calcium from decreased gastrointestinal absorption, PTH production increases in a counterregulatory attempt to release calcium from bone stores. Unfortunately, the accompanying phosphate release further exacerbates the hyperphosphatemia of CKD, which promotes further production of PTH.
The net result of these metabolic changes is a progressive and persistent increase in PTH levels. Although extremely high levels of PTH have been associated with worsened survival in patients with end-stage renal disease (ESRD) on hemodialysis, PTH and
bone minerals are not established surrogates of mortality. The principal association of hyperparathyroidism has been an increased risk of CKD-related bone disease.
bone minerals are not established surrogates of mortality. The principal association of hyperparathyroidism has been an increased risk of CKD-related bone disease.
BONE DISEASE AND CHRONIC KIDNEY DISEASE
The hyperparathyroidism of CKD has been most closely linked to the form of bone disease known as osteitis fibrosa cystica, characterized by markedly increased rates of bone turnover. Mineralization is preserved and bone volume is variable. The disease is often accompanied by bone marrow fibrosis, although a milder form of hyperparathyroid bone disease can also occur. The persistently high turnover seen in this disease is associated with a replacement of lamellar bone by woven bone, which is less resilient to stress. As a result, individuals with osteitis fibrosa cystica may be predisposed to fractures out of proportion to their bone density. While hyperparathyroidism is felt to be the strongest contributor to the development of osteitis fibrosa cystica, there may also be a contribution from 1,25-dihydroxyvitamin D deficiency itself.
The use of vitamin D replacement in CKD has been largely driven by the goal of preventing osteitis fibrosa cystica. Early studies demonstrated improvement in bone histology of osteitis fibrosa cystica after treatment with 25-hydroxyvitamin D. More recent studies have noted the ability of intravenous calcitriol to increase osteoblastic osteoid while reducing marrow fibrosis in patients on hemodialysis. Because of these findings, along with the physiology supporting suppression of PTH, vitamin D is considered to be an increasingly important therapy, particularly in patients on maintenance dialysis.
Increased use of vitamin D analogs may not come without risk, particularly with respect to bone disease. Adynamic bone disease, characterized by markedly reduced bone turnover, is the other major form of bone disease seen in CKD. Though mineralization is preserved, bone volume is usually low. As with osteitis fibrosa cystica, this disease has been linked to increased skeletal fragility. The prevalence of adynamic bone disease appears to be growing over time, especially in patients on peritoneal dialysis. This may be, at least in part, the result of increased use of active vitamin D analogs, as the development of adynamic bone disease has been reported in patients with osteitis fibrosa cystica treated with these agents.
The osteomalacia of CKD, characterized by a reduction in bone turnover and low to normal bone volume, is a third form of bone disease. Unlike adynamic bone disease, bone mineralization is markedly abnormal in osteomalacia. The distinguishing characteristic is an abundance of unmineralized osteoid, which is not typically seen in either adynamic bone disease or osteitis fibrosa cystica. The primary cause of osteomalacia is thought to be accumulation of aluminum and other heavy metals in bone, which can disrupt normal mineralization of the bone matrix. As aluminum-based phosphate binders have greatly declined in use, osteomalacia in CKD has faded from prominence.
MANAGING MINERAL METABOLISM IN CHRONIC KIDNEY DISEASE
The changes in calcium, phosphorous, and PTH that accompany CKD can be modified through the use of several therapeutic strategies, including an increasingly broad range of medications.
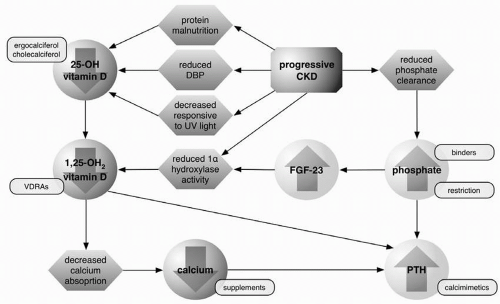 Figure 3-1. Changes in vitamin D and mineral metabolism associated with progressive chronic kidney disease. CKD, chronic kidney disease; FGF-23, fibroblast growth factor 23; PTH, parathyroid hormone; VDRA, vitamin D receptor antagonists. |
Phosphate Restriction and Binders
Phosphate control is a critical component in the management of CKD because of the protean effects of phosphate retention and hyperphosphatemia on a range of metabolic processes. As PTH promotes phosphate wasting, hyperphosphatemia can drive the development of hyperparathyroidism and osteitis fibrosa cystica. Phosphate retention promotes the production of the phosphatonin FGF-23, leading to suppression of 1α-hydroxylase and reduced production of 1,25-dihydroxyvitamin D (Fig. 3-1). Chronic elevation in serum phosphate, at least in patients with ESRD, also contributes to calcium phosphate deposition in tissues and vessel walls. In chronic hemodialysis patients, serum phosphorous levels above 6.5 mg/dL have been linked to an increased risk of early mortality. The importance of hyperphosphatemia in early stage CKD mortality is still a topic of investigation.
Given the wide-ranging deleterious effects of hyperphosphatemia, control of phosphate levels is an important focus of CKD management. Kidney Disease Outcomes Quality Initiative (K/DOQI) guidelines recommend maintaining serum phosphate between 2.7 mg/dL and 4.6 mg/dL in stage 3-4 CKD and between 3.5 mg/dL and 5.5 mg/dL in stage 5 CKD.
Dietary phosphate restriction is a reasonable initial approach with mild hyperphosphatemia, although it may have limited efficacy and practicability in some patients. K/DOQI guidelines recommend an intake of 800-1,000 mg daily for patients exceeding the target range for serum phosphate. Dairy products, beans, and meats are common sources of dietary phosphate but also important sources of protein. Thus, excessive focus on reducing dietary phosphorous could lead to inadvertent and excessive reduction in dietary protein. This may be particularly important in the dialysis
population, where nutritional status has been linked to patient outcomes. There are few studies directly evaluating the effects of dietary phosphate restriction alone on either serum phosphate or PTH levels. A detailed description of food and meal choices regarding phosphorus restriction is reviewed in Chapter 19.
population, where nutritional status has been linked to patient outcomes. There are few studies directly evaluating the effects of dietary phosphate restriction alone on either serum phosphate or PTH levels. A detailed description of food and meal choices regarding phosphorus restriction is reviewed in Chapter 19.
Related posts:
 Nutritional Support in Acute Renal Failure
Nutritional Support in Acute Renal Failure
 Requirements for Protein, Calories, and Fat in the Predialysis Patient
Requirements for Protein, Calories, and Fat in the Predialysis Patient
 Nutritional Requirements in Hemodialysis Patients
Nutritional Considerations in Patients on Peritoneal Dialysis
Nutritional Requirements in Hemodialysis Patients
Nutritional Considerations in Patients on Peritoneal Dialysis
 Exercise and Physical Function in Kidney Disease
Nutritional Interventions in Chronic Kidney Disease
Exercise and Physical Function in Kidney Disease
Nutritional Interventions in Chronic Kidney Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


