Fig. 63.1
Histological features of autoimmune hepatitis. a Portal and periportal lymphocyte and plasma cell infiltrate, extending to and disrupting the parenchymal limiting plate (interface hepatitis). Swollen hepatocytes, pyknotic necroses and acinar inflammation are present. Hematoxylin and eosin staining. b Reticulin staining of the same biopsy showing connective tissue collapse resulting from hepatocyte death and expanding from the portal area into the lobule (‘bridging collapse’)
There are three liver disorders in which liver damage is likely to arise from an autoimmune attack: autoimmune hepatitis (AIH), autoimmune sclerosing cholangitis (ASC) , and de novo AIH after liver transplant.
Autoimmune Hepatitis
Clinical Features
Two forms of AIH are recognized according to the type of autoantibody present in the serum (Table 63.1). Type 1 AIH (AIH-1) is positive for smooth muscle antibody (SMA) and/or antinuclear antibody (ANA), while type 2 AIH (AIH-2) for anti-liver/kidney microsomal type 1 (anti LKM-1) and/or anti-liver cytosol type 1 (anti-LC-1) antibody (Fig. 63.2) .
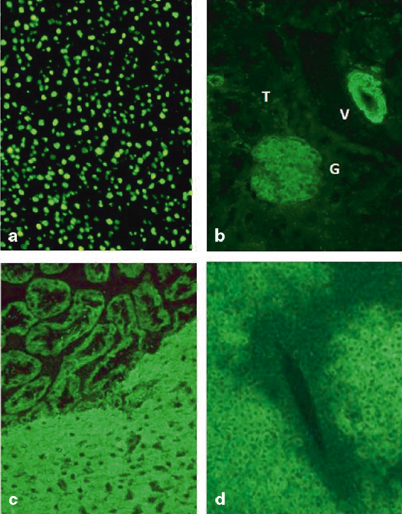
Fig. 63.2
Autoantibody immunofluorescence pattern on rodent tissue substrates. Panel A antinuclear antibody (ANA; liver): The homogeneous pattern is the most common in autoimmune hepatitis. Panel B smooth muscle antibody (SMA; kidney): SMA stains the smooth muscle of arterial vessels (V), glomeruli (G), and tubules (T; picture courtesy of Dr Luigi Muratori). Panel C anti-liver kidney microsomal type 1 antibody (LKM-1; kidney and liver): anti-LKM-1 stains the cytoplasm of hepatocytes and proximal renal tubules. Panel D anti-liver cytosol type 1 (anti-LC-1; liver): anti-LC-1 stains the cytoplasm of hepatocytes with a weakening of the stain around the central vein
Parameter | AIH-1 | AIH-2 | ASC |
|---|---|---|---|
Median age in years | 11 | 7 | 12 |
Mode of presentation (%) | |||
Acute hepatitis | 47 | 40 | 37 |
Acute liver failure | 3 | 25 | 0 |
Insidious onset | 38 | 25 | 37 |
Complication of chronic liver disease | 12 | 10 | 26 |
Associated autoimmune-disorders (%) | 22 | 20 | 48 |
Inflammatory bowel disease (%) | 20 | 12 | 44 |
Abnormal cholangiogram (%) | 0 | 0 | 100 |
ANA/SMA (%) | 100 | 25 | 96 |
Anti-LKM-1 (%) | 0 | 100 | 4 |
pANNA (%) | 45 | 11 | 74 |
Anti-SLA a (%) a | 58 | 58 | 41 |
Low C4 level (%) | 89 | 83 | 70 |
Increased frequency of HLA DR*0301 | Yes | Nob | No |
Increased frequency of HLA DR*0701 | No | Yes | No |
Increased frequency of HLA DR*1301 | No | No | Yes |
Interface hepatitis (%) | |||
Any degree | 92 | 94 | 60 |
Moderate/severe | 66 | 72 | 35 |
Histological biliary features (%) | 28 | 6 | 35 |
Cirrhosis (%) | 69 | 38 | 15 |
AIH-1 accounts for two thirds of the cases and presents often around puberty, while AIH-2 tends to present at a younger age and also during infancy. IgG is usually raised at disease onset in both types, though 15 % of children with AIH-1 and 25 % of those with AIH-2 have normal levels. IgA deficiency is common in AIH-2 [4]. Severity of disease is similar in the two types [4, 5], but anti-LKM1-positive children have higher levels of bilirubin and transaminases at onset than those who are ANA/SMA positive and present significantly more frequently with fulminant hepatic failure [4]. Excluding children with the fulminant presentation, a severely impaired hepatic synthetic function, as indicated by the presence of prolonged prothrombin time and hypoalbuminemia , is more common in AIH-1 than in AIH-2. The severity of interface hepatitis at diagnosis is similar in both types, but cirrhosis on initial biopsy is more frequent in AIH-1 than in AIH-2, suggesting a more chronic course of disease in the former. Progression to cirrhosis during treatment is more frequent in AIH-1.
In both types of AIH, a more severe disease course and a higher tendency to relapse are associated with the possession of antibodies to soluble liver antigen (SLA) [6, 7]. In both types, some 20 % of patients have associated autoimmune disorders—including thyroiditis, vitiligo, type 1 diabetes, inflammatory bowel disease (IBD), and nephrotic syndrome—and some 40 % have a family history of autoimmune disease (Table 63.1) [4].
a.
In at least 40 % of patients, the presentation is indistinguishable from that of an acute viral hepatitis (nonspecific symptoms of malaise, nausea/vomiting, anorexia, and abdominal pain, followed by jaundice, dark urine, and pale stools). Some children, particularly those who are anti-LKM1-positive, develop acute hepatic failure with grade II to IV hepatic encephalopathy (fulminant hepatitis) within 2–8 weeks from onset of symptoms.
b.
In 25–40 % of patients, the onset is insidious, with an illness characterized by progressive fatigue, relapsing jaundice, headache, anorexia, amenorrhea, and weight loss, lasting for several months and even years before diagnosis.
c.
In about 10 % of patients, there is no history of jaundice, and the diagnosis follows presentation with complications of portal hypertension, such as splenomegaly, haematemesis from oesophageal varices , bleeding diathesis, chronic diarrhea, and weight loss.
The mode of presentation of AIH in childhood is therefore variable, and the disease should be suspected and excluded in all children presenting with symptoms and signs of liver disease not ascribable to more common pathologies. The course of the disease can be fluctuating, with flares and spontaneous remissions, a pattern that may result in delayed referral and diagnosis. The majority of children, however, on physical examination have clinical signs of an underlying chronic liver disease, including cutaneous stigmata (spider nevi, palmar erythema, leukonychia, striae), firm liver, and splenomegaly. At ultrasound, the liver parenchyma of these patients is often nodular and heterogeneous.
Epidemiology and Genetic Predisposition
The epidemiology of childhood AIH has not been studied. Data collected at the King’s College Hospital Pediatric Hepatology tertiary referral centre show an increase in the yearly incidence of juvenile autoimmune liver disease, only partially explained by a referral bias: In the 1990s, it represented 2.3 % of some 400 children older than 4 months who were newly referred yearly; since 2000, the yearly incidence has increased to 12 %.
In northern Europe, pediatric AIH-1, similar to adult AIH, is associated with the possession of the human leukocyte antigen (HLA) DRB1*03 (Table 63.1) [4, 8]. In contrast to adult patients, possession of DRB1*04 does not predispose to AIH in childhood, and this can even exert a protective role [4]. AIH-2 is associated with possession of DRB1*07, and, in DR7-negative patients, with possession of DRB1*03 (Table 63.1) [9, 10]. In Egypt, AIH-2 appears to be associated also with possession of HLA-DRB1*15 [11]. In Brazil and Egypt, the primary susceptibility allele for AIH-1 is DRB1*1301, but a secondary association with DRB1*0301 has also been identified [11, 12]. Interestingly, in South America, possession of the HLA-DRB1*1301 allele not only predisposes to pediatric AIH-1 but is also associated with persistent infection with the endemic hepatitis A virus [13, 14]. Pediatric patients with AIH, whether anti-LKM-1 or ANA/SMA positive, have isolated partial deficiency of the HLA class III complement component C4, which is genetically determined [15].
Diagnosis
Liver biopsy is necessary to establish the diagnosis; the typical histological picture including a dense mononuclear and plasma cell infiltration of the portal areas, which expands into the liver lobule, destruction of the hepatocytes at the periphery of the lobule with erosion of the limiting plate (‘interface hepatitis’; Fig. 63.1a), connective tissue collapse resulting from hepatocyte death and expanding from the portal area into the lobule (‘bridging collapse’; Fig. 63.1b), and hepatic regeneration with ‘rosette’ formation. In addition to the typical histology , other positive criteria include elevated serum transaminase and IgG levels and presence of ANA, SMA, anti-LKM-1, or anti-LC-1.
The diagnosis of AIH has been advanced by the scoring systems developed by the International Autoimmune Hepatitis Group (IAIHG) for adult patients [1, 2] where negative criteria, such as evidence of infection with hepatitis B or C virus, Wilson disease, or alcohol, among others, are taken into account in addition to the positive criteria mentioned above. The IAIHG scoring system was devised mainly for research purposes to allow ready comparison between series from different centres, but it has also been used clinically, including in pediatric series. More recently, the IAIHG has published a simplified scoring system based on autoantibodies, IgG, histology, and exclusion of viral hepatitis that is better suited to clinical application [20]. However, neither scoring system is suitable to the juvenile form of the disease, where diagnostically relevant autoantibodies often have titers lower than the cut-off value considered positive in adults [21–23]. In addition, neither system can distinguish between AIH and ASC (see below) [24, 25], which can only be differentiated if a cholangiogram is performed at presentation .
A key diagnostic criterion for all AIH scoring systems is the detection of autoantibodies (ANA, SMA, anti-LKM-1, and anti-LC-1), which not only assists in the diagnosis but also allows differentiation of AIH types. ANA and SMA that characterize AIH-1 and anti-LKM-1 and anti-LC-1 that define AIH-2 are usually mutually exclusive; in those instances when they are present simultaneously, the clinical course is similar to that of AIH-2 [26]. A major target of SMA is the actin of smooth muscle, whereas the molecular target of anti-LKM-1 is cytochrome P-4502D6 (CYP2D6) [27] and of anti-LC-1 is formiminotransferase cyclodeaminase [28]. ANA, SMA, anti-LKM-1, and anti-LC-1 should be sought by indirect immunofluorescence using rodent stomach, kidney, and liver as substrate, as other techniques, for example commercially available enzyme-linked immunosorbent assay (ELISA), remain to be fully validated (Fig. 63.2) [26]. In contrast to adults, in healthy children, autoantibody reactivity is infrequent, so that titers of 1/20 for ANA and SMA and 1/10 for anti-LKM-1 are clinically relevant. Anti-LC-1, also detectable by indirect immunofluorescence, can be present on its own, but frequently occurs in association with anti-LKM-1. This co-occurrence can go unnoticed because anti-LKM-1 obscures the anti-LC-1 pattern. Anti-LC-1 can also be detected by commercial ELISA. Positivity for autoantibodies is not sufficient for the diagnosis of AIH since they can be present, usually at low titer, in other liver disorders such as viral hepatitides [29, 30], Wilson disease [31], and nonalcoholic steatohepatitis [32] .
Other autoantibodies less commonly tested but of diagnostic importance include peripheral antinuclear neutrophil antibody (atypical perinuclear anti-neutrophil cytoplasmic antibodies, pANCA or peripheral anti-nuclear neutrophil antibodies, pANNA) and anti-SLA. pANNA is frequently found in AIH-1 and ASC, and it is also common in IBD, while it is virtually absent in AIH-2. Anti-SLA, originally described as the hallmark of a third type of AIH [33], is also found in up to 50 % of patients with AIH-1, AIH-2, or ASC, where it defines a more severe course [6, 7]. Anti-SLA is not detectable by immunofluorescence, but the definition of its molecular target as Sep (O-phosphoserine) tRNA:Sec (selenocysteine) tRNA synthase (SepSecS) [34, 35] has enabled the establishment of molecularly based diagnostic assays. However, it should be noted that ELISAs are less sensitive than radioligand assays available in research laboratories [34, 35].
There are a small proportion of patients with AIH without detectable autoantibodies. This condition, which responds to immunosuppression like the seropositive form, represents seronegative AIH [36], a rare type of AIH in adults, whose prevalence and clinical characteristics remain to be defined in children .
Pathophysiology
The typical histologic picture of AIH, which is characterized by a dense mononuclear cell infiltrate eroding the limiting plate and invading the parenchyma (interface hepatitis, Fig. 63.1a), first suggested that auto-aggressive cellular immunity might be involved in its causation [37]. Immunocytochemical studies have identified the phenotype of the infiltrating cells. T lymphocytes mounting the alpha/beta T cell receptor predominate. Among the T cells, a majority is positive for the cluster of differentiation (CD)4 helper/inducer phenotype, and a sizable minority is positive for the CD8 cytotoxic phenotype. Lymphocytes of non-T cell lineage are fewer and include (in decreasing order of frequency) natural killer cells (CD16/CD56 positive), macrophages, and B lymphocytes [38]. Natural killer T cells, which express simultaneously markers of both natural killer (CD56) and T cells (CD3), are involved in liver damage in an animal model of AIH [39].
A powerful stimulus must be promoting the formation of the massive inflammatory cell infiltrate present at diagnosis. Whatever the initial trigger, it is most probable that such a high number of activated inflammatory cells cause liver damage. There are different possible pathways that an immune attack can follow to inflict damage on the hepatocyte (Fig. 63.3). Liver damage is believed to be orchestrated by CD4-positive T lymphocytes recognizing a self-antigenic peptide. To trigger an autoimmune response, the peptide must be embraced by an HLA class II molecule and presented to uncommitted T helper (Th0) cells by professional antigen-presenting cells (APCs), with the co-stimulation of ligand–ligand (CD28 on Th0, CD80 on APC) interaction between the two cells. The Th0 cells become activated, differentiate into functional phenotypes according to the cytokines prevailing in the microenvironment and the nature of the antigen, and initiate a cascade of immune reactions determined by the cytokines they produce. Arising in the presence of the macrophage-produced interleukin-12 (IL-12), Th1 cells secrete mainly IL-2 and interferon-γ, which activate macrophages, enhance expression of HLA class I (increasing the vulnerability of liver cells to cytotoxic attack), and induce expression of HLA class II molecules on hepatocytes, which then become able to present the autoantigenic peptide to Th cells, thus perpetuating the immune recognition cycle. Th2 cells, which differentiate from Th0 if the microenvironment is rich in IL-4, produce mainly IL-4, -5, and -10, which induce autoantibody production by B lymphocytes. Physiologically, Th1 and Th2 cells antagonize each other. The process of autoantigen recognition is strictly controlled by regulatory mechanisms. If these regulatory mechanisms fail, the autoimmune attack is perpetuated. Over the past three decades, different aspects of the above pathogenic scenario have been investigated.
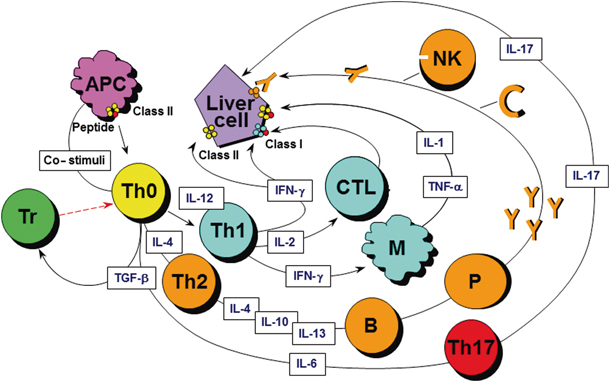
Fig. 63.3
An autoantigen is presented to uncommitted T helper (Th0) lymphocytes within the HLA class II molecule of an antigen-presenting cell (APC) in either the regional lymph nodes or within the liver itself. Activated Th0 cells differentiate into Th1 or Th2 cells in the presence of interleukin (IL)-12 or IL-4 and according to the nature of the antigen, and trigger a series of immune reactions determined by the cytokines they produce: Th1 cells secrete IL-2 and interferon-gamma (IFN-γ), cytokines that stimulate cytotoxic T-lymphocytes (CTL), enhance expression of class I and induce expression of class II HLA molecules on the liver cells and activate macrophages. The latter release IL-1 and tumour necrosis factor-alpha (TNF-α). Th2 cells secrete mainly IL-4, IL-10, and IL-13, and stimulate autoantibody production by B lymphocytes. In the presence of defective regulatory T cells (Treg), hepatocyte destruction ensues from the engagement of damaging effector mechanisms, including CTL, cytokines released by Th1, and by activated macrophages, complement activation, or adhesion of natural killer (NK) cells to autoantibody-coated hepatocytes through their Fc receptors. Th17 cells, a recently described proinflammatory population that derives from Th0 cells in the presence of transforming growth factor beta (TGF-β) and IL-6, are the focus of current investigation. (Adapted with permission from Ref. [40], with permission from Wiley-Blackwell)
An impairment of immunoregulatory mechanisms has been described in AIH. Both children and young adults with this condition have low levels of T cells expressing the CD8 marker and impaired suppressor cell function [41, 42] which segregate with the possession of the HLA haplotype B*08/DRB1*03 (formerly B8/DR3), and can be corrected by therapeutic doses of corticosteroids [43]. Furthermore, patients with AIH have been reported to have a specific defect in a subpopulation of T cells controlling the immune response to liver-specific membrane antigens [44]. Further evidence for an impairment of immunoregulatory function in AIH has been obtained in the past decade [45–47]. Among T cell subsets with potential immunosuppression function, CD4+ T cells constitutively expressing the IL-2R alpha chain (CD25; regulatory T cells, T-regs) have emerged as the dominant immunoregulatory population [48]. These cells, which represent 5–10 % of the total population of peripheral CD4+ T lymphocytes, control the innate and the adaptive immune responses by preventing the proliferation and effector function of autoreactive T cells. Their mechanism of action involves mainly a direct contact with the target cells, and, to a lesser extent, the release of immunoregulatory cytokines, such as IL 10 and tissue growth factor beta 1.
A numerical T-reg impairment affects both children and adults with AIH. This defect is more evident at diagnosis than during drug-induced remission, although even then circulating T-reg frequencies fail to reach the levels seen in health [45, 49, 50]. The percentage of T-regs inversely correlates with biomarkers of disease severity, suggesting that a reduction in regulatory T cells favours autoimmune liver disease. Importantly, T-regs from AIH patients at diagnosis are impaired in their ability to control the proliferation of CD4 and CD8 effector cells compared to T-regs isolated from AIH patients at remission or healthy subjects [49].
Recently, it was reported that effector CD4 T cells isolated from patients with AIH are less susceptible to the regulatory control exerted by T-regs. This defect is linked to reduced expression of the inhibitory receptor T cell immunoglobulin- and mucin-domain-containing-molecule-3 (Tim-3), which upon ligation of galectin-9 expressed by T-regs, induces effector cell death [51].
Hepatocytes from patients with AIH, in contrast to normal hepatocytes, express HLA class II molecules [52], and, although lacking the antigen-processing machinery typical of APCs, they may present peptides through a bystander mechanism. Given the impaired regulatory function and the inappropriate expression of HLA class II antigens on the hepatocytes, it is conceivable that an autoantigenic peptide is presented to the helper/inducer cells, leading to their activation. Although there is no direct evidence as yet that an autoantigenic peptide is presented and recognized, activation of helper cells has been documented in AIH [38, 53]. These activated cells possess the CD4 phenotype, and their numbers are highest when the disease is most active.
Most advances in the study of T cells have occurred in AIH-2, since the knowledge that CYP2D6 is the main autoantigen has enabled the characterization of both CD4 and CD8 T cells targeting this cytochrome. One study has shown that CD4 T cells from patients with AIH-2 positive for the predisposing HLA allele DRB1*0701 recognize seven regions of CYP2D6 [9], five of which have later been shown to be also recognized by CD8 T cells [54]. A high number of interferon-gamma-producing CD4 T cells and CD8 T cells are associated with biochemical evidence of liver damage, suggesting a combined cellular immune attack.
What triggers the immune system to react to an autoantigen is unknown. A lesson may be learned by the study of humoral autoimmune responses during viral infections. Thus, studies aimed at determining the specificity of the LKM-1 antibody, present in both the juvenile form of AIH and some patients with chronic hepatitis C virus (HCV) infection, have shown a high amino acid sequence homology between the HCV polyprotein and CYP2D6, the molecular target of anti-LKM-1, thus implicating a mechanism of molecular mimicry as a trigger for the production of anti LKM-1 in HCV infection [27, 55, 56]. It is therefore conceivable that an as-yet unknown virus infection may be at the origin of the autoimmune attack in AIH.
Titers of antibodies to liver-specific lipoprotein, a macromolecular complex present on the hepatocyte membrane, and its well-characterized component asialoglycoprotein receptor, correlate with the biochemical and histologic severity of AIH [57, 58]. Antibodies to alcohol dehydrogenase, a second well-defined component of liver-specific lipoprotein, have been described in patients with AIH [59]. Immunofluorescence studies on monodispersed suspensions of liver cells obtained from patients with AIH showed that these cells are coated with antibodies in vivo [60]. A pathogenic role for these autoantibodies has been indicated by cytotoxicity assays showing that autoantibody-coated hepatocytes from patients with AIH are killed when incubated with autologous lymphocytes. The effector cell was identified as an Fc receptor-positive mononuclear cell [61]. T cell clones obtained from liver biopsies of children with AIH and expressing the gamma/delta T cell receptor have been shown to be cytotoxic to a variety of targets but preferentially kill liver-derived cells as opposed to cell lines derived from other organs [62].
The establishment of cell lines and clones has shown that the majority of T cell clones obtained from the peripheral blood and a proportion of those from the liver of patients with AIH are CD4 positive and use the conventional alpha/beta T cell receptor [62–65]. Some of these CD4-positive clones were further characterized and were found to react with partially purified antigens, such as crude preparations of liver cell membrane or liver-specific lipoprotein [63], and with purified asialoglycoprotein receptor [63, 65] or recombinant CYP2D6 [64] and to be restricted by HLA class II molecules in their response. Because CD4 is the phenotype of Th cells, T cell clones were investigated for their ability to help autologous B lymphocytes in the production of immunoglobulin in vitro [63, 65]. Indeed, their coculture with B lymphocytes resulted in a dramatic increase in autoantibody production.
The possible role of Th17 cells in the pathogenesis of AIH is under investigation. Th17 cells contribute to autoimmunity by producing the proinflammatory cytokines IL-17, IL-22, and TNF-α and inducing hepatocytes to secrete IL-6 [66], which further enhances Th17 activation. Th17 cells have been shown to be elevated in the circulation and liver of patients with AIH [66].
Treatment
Unless it presents with fulminant hepatic failure (i.e. acute liver failure and encephalopathy, which usually requires urgent transplantation), AIH responds satisfactorily to immunosuppression, even in the presence of poor synthetic function and/or established cirrhosis. Treatment should be started with prednisolone 2 mg/g/day (maximum 60 mg/day), which is gradually decreased over a period of 4–8 weeks if there is progressive normalization of the transaminases, and then the patient is maintained on the minimal dose able to sustain normal transaminase levels, usually 5 mg/day. During the first 6–8 weeks of treatment, liver function tests should be checked weekly to allow a frequent fine-tuning of the treatment, avoiding severe steroid side effects. The initial goal is to obtain at least 80 % of reduction of the transaminase levels by 8 weeks of treatment. During this period of time, if progressive normalization of the liver function is not observed at weekly blood tests, azathioprine is added at a starting dose of 0.5 mg/g/day, which, in the absence of signs of toxicity, is increased up to a maximum of 2–2.5 mg/g/day until remission (i.e. normal transaminase levels) is achieved. Azathioprine is not recommended as first-line treatment because of its potential hepatotoxicity, particularly in severely jaundiced patients. Interestingly, it has been shown that neither thiopurine methyltransferase genotype nor activity predicts azathioprine hepatotoxicity in AIH, which appears instead to be related to the degree of liver fibrosis [67]. A preliminary report in a cohort of 30 children with AIH suggests that the measurements of the azathioprine metabolites 6-thioguanine and 6-methylmercaptopurine are useful in identifying drug toxicity and non-adherence and in achieving a level of 6-thioguanine considered therapeutic for IBD [68]. However, it has been reported that patients with AIH can achieve remission with azathioprine metabolite levels lower than those needed for IBD [69].
Although an 80 % decrease of initial transaminase levels is usually obtained within 6–8 weeks from starting treatment in most patients, complete normalization of liver function may take several months. In our own series, normalization of transaminase levels occurred at medians of 0.5 years (range 0.2–7 years) in ANA/SMA-positive children and 0.8 years (range 0.02–3.2 years) in anti-LKM-1-positive children [4]. Relapse while on treatment is common, affecting about 40 % of the patients and requiring a temporary increase of the steroid dose. An important role in relapse is played by non-adherence, particularly in adolescents [70]. The risk of relapse is higher if steroids are administered on an alternate-day schedule, often instituted in the unsubstantiated belief that it has a less negative effect on the child’s growth. Small daily doses should be used because they are more effective in maintaining disease control and minimize the need for high-dose steroid pulses during relapses (with attendant more severe side effects). Side effects of steroid treatment are usually mild, the only serious complication being psychosis during induction of remission in 4 %, which resolves after prednisolone withdrawal [4]. All patients develop a transient increase in appetite and mild cushingoid features during the first few weeks of treatment. After 5 years of treatment, 56 % of our patients maintained their baseline centile for height or went up across a centile line, 38 % dropped across one centile line, and only 6 % dropped across two centile lines [71]. Moreover, all patients achieved their expected final height after a median steroid treatment of 8.9 years [72].
Treatment should be continued for at least 3 years before considering its cessation, after which period stopping treatment can be attempted but only if liver function tests and IgG levels have been persistently normal, and autoantibodies are either undetectable or detectable at very low titre (ANA/SMA < 1:10; anti-LKM-1 should be negative) over at least 12 months, and a follow up liver biopsy shows no inflammatory changes. However, it is advisable not to attempt treatment withdrawal during or immediately before puberty, when relapses are more common. Treatment withdrawal is reportedly successful in 20 % of children with AIH-1, but is rarely achieved in AIH-2 [4]. An important role in monitoring the response to treatment is the measurement of autoantibody titers and IgG levels, the fluctuation of which is correlated with disease activity [73]. The prognosis of those children with AIH who respond to immunosuppressive treatment is generally good, with most patients surviving long term with excellent quality of life on low-dose medication. Development of end-stage liver disease requiring liver transplantation despite treatment, however, has been reported 8–14 years after diagnosis in 8.5 % of children with AIH [4].
Maintenance with azathioprine monotherapy has been advocated once remission is achieved [74], but whether this is effective long term and whether it offers any benefit on possible side effects compared to low-dose prednisolone/azathioprine maintenance is unclear.
Induction of remission has been obtained in 72 % treatment-naïve children with AIH-1 using cyclosporine A alone for 6 months, followed by maintenance with low-dose prednisone and azathioprine [75]. A 5-year follow-up of this study shows that 94 % of the patients eventually achieved remission, with minor side effects [76]. Whether this mode of induction has any advantage over the standard treatment remains to be evaluated in controlled studies in specialized centres.
Tacrolimus is a more potent immunosuppressive agent than cyclosporine, but it also has significant toxicity. There is limited evidence supporting its role as initial treatment of AIH apart from anecdotal reports in adults.
Budesonide has a hepatic first-pass clearance of > 90 % of oral dose and fewer side effects than predniso(lo)ne, but cannot be used in the presence of cirrhosis, which affects at least two thirds of AIH patients. In a large European study, including adults and children with AIH, a combination of budesonide and azathioprine had fewer adverse effects compared to medium-dose standard prednisone and azathioprine [77]. In this study, budesonide at a dose of 3 mg three times daily, decreased upon response, was compared with prednisone 40 mg once daily reduced per protocol irrespective of response. After 6 months of treatment, remission was achieved in 60 % of the budesonide group but in only 39 % of the prednisone group, both percentages being worse than those achieved with standard treatment [4]. The results among the children recruited into this study were particularly disappointing, with a similarly low remission rate of 16 % for budesonide and 15 % for prednisone after 6 months of treatment and of 50 and 42 %, respectively, after 12 months of treatment, with similar steroid side effects in both groups, apart from higher frequency of weight gain in children on prednisone [78]. Large controlled studies are needed to establish the appropriate dose for children [79]. Nevertheless, budesonide could be a valid alternative in selected non-cirrhotic patients who are at risk of adverse effects from steroids.
In those patients (up to 10 %) in whom standard immunosuppression is unable to induce stable remission, or who are intolerant to azathioprine, mycophenolate mofetil at a dose of 20 mg/kg twice daily has been successfully used [80]. In case of persistent no response or of intolerance to mycophenolate mofetil (headache, diarrhea, nausea, dizziness, hair loss and neutropenia), the use of calcineurin inhibitors (cyclosporine A or tacrolimus) should be considered.
Children who present with fulminant hepatic failure, that is with grade 3–4 encephalopathy and international normalized ratio (INR) ≥ 2, pose a particularly difficult therapeutic problem. Although it has been reported that they may benefit from conventional immunosuppressive therapy [81, 82], most require liver transplantation [4]. Encouraging results have been reported using cyclosporine A in anti-LKM-1-positive patients presenting with fulminant hepatitis [81]. These should be evaluated on a larger number of patients because our own experience has not confirmed the value of this therapeutic approach.
Autoimmune Polyendocrinopathy–Candidiasis–Ectodermal Dystrophy (APECED)
APECED is a monogenic disorder due to mutations of the AIRE1 gene [16, 17]. Its phenotype is variable and includes AIH in about 20 % of the cases [19]. This resembles AIH-2 and responds to standard immunosuppressive treatment [18], though the anti-LKM-1-like antibody detected by immunofluorescence targets a cytochrome (P4502A6) different from that of classical AIH-2. APECED, also known as autoimmune polyendocrine syndrome 1, is an autosomal recessive disorder caused by homozygous mutations in the AIRE1 gene and characterized by a variety of organ-specific autoimmune diseases, the most common of which are hypoparathyroidism and primary adrenocortical failure, accompanied by chronic mucocutaneous candidiasis.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


