Approach to the Diagnosis of Glomerular Disease
A. Neil Turner
The current nomenclature for glomerulonephritis is a confusing mixture of terms derived from light microscopy and those derived from more detailed investigations; for example, focal segmental glomerulosclerosis (FSGS), membranoproliferative glomerulonephritis (MPGN), and crescentic nephritis are all descriptions based on light microscopy. However, the labels of immunoglobulin (Ig) A nephropathy, amyloidosis, or anti-glomerular basement membrane (GBM) disease are all applied on the basis of additional information about specific molecules identified using supplementary techniques. Other diagnoses are based on serologic, and, sometimes also, genetic tests. These appear to give further information about pathogenesis, but for most conditions, there remains much more to learn.
Even before kidney biopsy is performed, simple clinical findings provide clues to both the presence of glomerulonephritis and to the nature of the pathologic process behind it.
RESPONSES TO INJURY IN THE GLOMERULUS
The glomerulus has a highly refined and beautiful structure to perform its functions. Its key components are few: three main cell types (Figure 3.1) plus the GBM. Its responses to injury are quite limited, and looking at them gives a strong steer to causation. The three major manifestations of glomerulonephritis, of which you can have one or several, are as follows:
Loss of filtration capacity
Protein leak (proteinuria)—podocyte problem
Blood leak (hematuria)—GBM damage
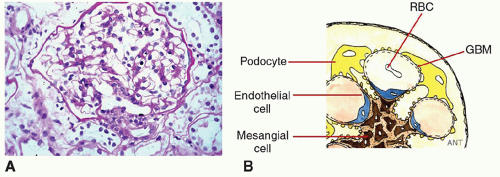 FIGURE 3.1: The glomerulus. A, Normal glomerulus. (Courtesy of Dr Chris Bellamy.) B, Cartoon of three glomerular capillary loops showing cell types. GBM, glomerular basement membrane; RBC, red blood cell. |
There are two important, additional but inconsistent, manifestations of glomerulonephritis that we return to later in the chapter: hypertension and fluid retention.
The mechanisms of proteinuria and hematuria are quite different, and this allows separation of the causes of glomerulonephritis.
PROTEINURIA AS A PODOCYTE ISSUE
Generally speaking, proteinuria is caused by podocyte dysfunction. The evidence for this is strong. Genetic causes of nephrotic-level proteinuria are mutations that affect the podocyte structure or function or pertain to molecules that interact with podocytes. Where we understand the pathogenesis of primary kidney diseases, the podocyte is the target of those in which proteinuria is a primary feature.
Where proteinuria is a secondary feature of glomerular diseases, it is associated with disordered composition of the matrix beneath the podocyte and/or disordered architecture. A “hostile environment” in which the podocyte fails to receive the usual signals that cause it to maintain the filtration barrier leads to proteinuria.
PROTEINURIA AS A LATE CONSEQUENCE OF GLOMERULAR DAMAGE
The normal structure and composition of the GBM is critical for podocytes to be able to conserve the filtration barrier. Although some glomerular diseases resolve almost completely, many leave scarring, with altered architecture and abnormal protein deposition. This disrupts podocyte differentiation and function and is associated with proteinuria.
Proteinuria is quantitatively associated with the long-term risk of progressive loss of kidney function. The mechanisms of progressive loss of glomerular filtration rate (GFR) after an episode of acute glomerulonephritis or time-limited glomerular injury are not entirely elucidated. Is the proteinuria the mediator of further damage or just a marker of distressed podocytes?
DETAILED CLINICAL ASSESSMENT OF PROTEINURIA
Proteinuria comes from the kidney, with the following rare exceptions:
Chyluria
Severe exudative cystitis
Intestinal tissue after fashioning of a urinary conduit
Deliberate adulteration of the urine sample
The quantitation of proteinuria is valuable not only in determining long-term prognosis (all specialties) but also (for nephrologists) in determining its origin, glomerular versus tubular (Figure 3.2). Quantitation is achieved by relating excretion to time (24-hour collection) or to excretion of creatinine (spot samples for urine protein:creatinine ratio [UPCR] or urine albumin:creatinine ratio [UACR]). Spot measures have the advantage of being easy to repeat and monitor long term. In research studies, the results can be more variable than are high-quality 24-hour collections, but in the real world, it is hard to repeat accurate collections frequently. In either case, it would be wrong to base major management decisions on a single result from either test. In clinical practice, one should generally follow trends based only on the same type of measurements (ie, either sequential UPCR/UACR or 24-hour protein excretion).
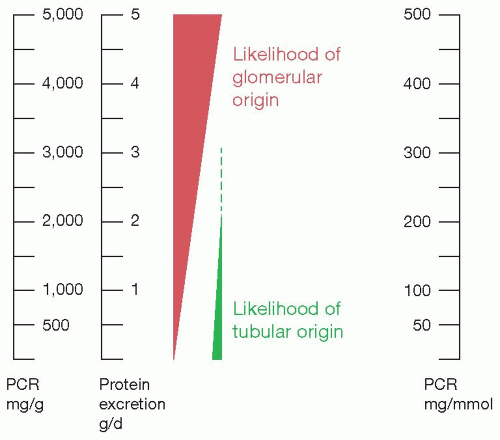 FIGURE 3.2: Proteinuria versus likelihood of glomerular versus tubular disease. PCR, protein: creatinine ratio. |
Figure 3.2 shows how the likelihood of glomerular disease relates to the quantity of protein. The amount of low-molecular-weight filtered tubular protein is limited, so that higher quantities of total protein are increasingly likely to be glomerular in origin, particularly if more than 2 g/d. The only exceptions are where abnormal quantities of low-molecular-weight proteins are synthesized and excreted. This is seen most commonly in myeloma, with overproduction of Ig light chains, and exceptionally rarely, lysozyme is overproduced in monomyelocytic leukemia.
Glomerular protein has a high content of serum albumin (molecular mass 67 kDa), whereas tubular proteins are filtered at the normal glomerulus and so have molecular mass usually below 30 kDa. Low-molecular-weight proteins are occasionally assayed as a measure of the failure of tubular reabsorption, or an electrophoretic gel can show size distribution, but the easiest test is to measure both albumin and total protein in the same urine sample (so, both albumin:creatinine ratio [ACR] and protein:creatinine ratio [PCR]). If albumin is more than 40% of the total, the proteinuria is likely to be glomerular.1,2
Very heavy proteinuria is a cardinal feature of nephrotic syndrome (see Chapter 4). The causes are glomerular diseases at the proteinuric end of the spectrum.
PITFALLS IN ESTIMATING PROTEINURIA
Fever and Other Illnesses
Acute illness and fevers, and heart failure, are associated with increased protein excretion that quickly returns to normal.
Pregnancy
The upper limit of normal for ACR and PCR is increased in pregnancy. Pregnancy may amplify levels of proteinuria in patients with glomerular disease, sometimes severely enough to cause nephrotic syndrome (see Chapter 22).
Postural Proteinuria
Postural or orthostatic proteinuria is mainly described as a normal developmental phenomenon in children.3 It may be quite extreme, with protein excretion rates during the daytime that falls well into the nephrotic range, but with normal values on a first-in-morning sample. The way to avoid overdiagnosis is always to use first-in-morning samples in children. These should contain normal levels of protein.
There is not a defined age at which postural proteinuria disappears, and it may also be seen in young adults. Some young patients with glomerular disease can show marked variation in proteinuria levels during the day, so early morning samples are likely to give more consistent results if monitoring glomerular disease.
Exertional Proteinuria and Hematuria
Vigorous short-term exercise is associated with transient proteinuria. Prolonged running or marching is also associated with hematuria; see subsequent text.
Hypertension
Hypertension alone should not be considered an adequate explanation for the presence of proteinuria, except in malignant (accelerated phase) hypertension. However, the proteinuria would usually justify investigation for underlying glomerular disease on its own.
Severe hypertension increases proteinuria levels in patients with underlying glomerular disease. This is the converse of the observation that lowering blood pressure reduces levels of proteinuria.
HEMATURIA IS CAUSED BY DISRUPTION OF THE GLOMERULAR BASEMENT MEMBRANE
A disruption of the integrity of the GBM must be present for red cells to cross the glomerular capillary wall into the Bowman space and into the urine, resulting in glomerular hematuria. As with proteinuria, GBM weakness can be caused by inherited diseases—see Alport syndrome and other collagen disorders, Chapter 21. But most commonly, the holes are caused by inflammation, by direct or bystander injury in an immune response. In these conditions, you may see antibody deposition, but you also see cells of the immune system (lymphocytes, macrophages) invading the glomerulus.
You can also see the attempts of the glomerulus to recover from injury, with proliferation of intrinsic cells in capillary loops (endothelial cells) or mesangium.
Additional immune and native cells often lead to a hypercellular appearance, and these may be called proliferative types of glomerulonephritis.
Hematuria can arise anywhere in the urinary tract, and also, on occasion, from interstitial diseases. See the following for details on the clinical assessment of hematuria.
DETAILED CLINICAL ASSESSMENT OF HEMATURIA
In contrast to proteinuria, hematuria commonly has its origin elsewhere in the urinary tract. This is of particular concern in older patients or in those with risk factors for urothelial malignancy. Malignancy is particularly likely as an explanation in the presence of visible hematuria.
Visible Hematuria
For patients with visible hematuria, guidelines recommend urologic investigation as the first step for those over a threshold age, for example, 40 years, in the absence of strong pointers to intrinsic kidney disease.
Pointers to kidney disease include
Impaired and particularly deteriorating GFR
Proteinuria
Hypertension
Dysmorphic red blood cells (RBCs) or RBC casts seen on microscopic examination of the spun urine sediment
Although the frequency of hypertension, decreased GFR, and proteinuria increase with age, their presence should not delay urologic investigation if the risk of quickly progressive kidney disease is shown to be low. Conversely, marked proteinuria or declining kidney function is an indication for the need of a prompt assessment of glomerulonephritis, which should not be delayed to complete a urologic evaluation.
Microscopic Hematuria
Microscopic (invisible) hematuria is common, and the likelihood of malignancy explaining it is much lower. In young patients with persistent microscopic hematuria but no recognized underlying conditions or comorbidity, the presence of any proteinuria, even microalbuminuria, is a strong pointer to glomerular disease as the cause. In older or comorbid populations, the high background incidence of hypertension and microalbuminuria reduces the reliability of these features.
Other Kidney Causes of Hematuria
Hematuria may also be seen in acute tubulointerstitial inflammation, including pyelonephritis. When used as a single indicator, it is therefore much less specific a finding than is proteinuria in establishing a diagnosis of glomerular disease, or, indeed, kidney disease. Nevertheless, on a population basis, either or both hematuria and proteinuria are associated with an increased rate of end-stage kidney disease, presumably through association with glomerulonephritis.
Hematuria is a recognized manifestation of sickle cell disease and trait, thought to be mediated by medullary ischemia. Papillary necrosis may occur and cause overt bleeding and even urinary obstruction. Other causes of papillary necrosis can do the same.
URINE MICROSCOPY
Examination of urinary sediment by phase-contrast microscopy can be useful in distinguishing glomerular hematuria from other causes, by visualization of red cell casts or of red cells that have been fragmented or deformed by squeezing through breaks in the GBM. Acanthocytes, in which polydiverticular pear-shaped extrusions protrude from ring-shaped erythrocytes, are quite specific for glomerular bleeding if strictly defined (Figure 3.3).4,5 There are a number of keen advocates for increasing the use of this technique in clinical practice, but the requirement for significant skilled time, equipment, and space to achieve reliable results has disappointingly limited wide adoption.
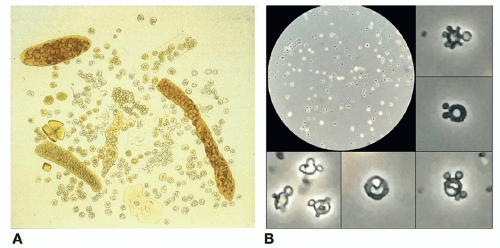 FIGURE 3.3: Glomerular red cells on urine microscopy. A, Red cell cast (From Atlas of Urinary Sediments: With Special Reference to Their Clinical Significance/by Hermann Rieder; translated by Frederick Craven Moore; edited and annotated by A. Sheridan Delépine.) B, Acanthocytes by phase-contrast microscopy (Courtesy of Dr Florian Buchkremer.). FSGS, focal segmental glomerulosclerosis; GBM, glomerular basement membrane; IgA, immunoglobulin A; MPGN, membranoproliferative glomerulonephritis; SLE, systemic lupus erythematosus. |
PITFALLS IN ESTIMATING HEMATURIA
The main concern is distinguishing hematuria arising elsewhere in the urinary tract, or in other intrarenal conditions, including stones and tumors.
Strenuous Exercise-Related Hematuria and/or Proteinuria
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


