CHAPTER 108 Antibiotic-Associated Diarrhea, Pseudomembranous Enterocolitis, and Clostridium difficile-Associated Diarrhea and Colitis
ANTIBIOTIC-ASSOCIATED DIARRHEA
ETIOLOGY
Diarrhea is a common side effect of antibiotic use and can result from a variety of mechanisms.1 The most common type of diarrhea, often called simple antibiotic-associated diarrhea (AAD), is believed to result from a disturbance of the normal colonic microflora, leading to alterations in bacterial degradation of nonabsorbed carbohydrates and bile salts. Colonic bacteria normally ferment the complex carbohydrates in dietary fiber and other carbohydrates that are not absorbed in the small intestine, and the fermentation products are then metabolized and absorbed in the colon. Disruption of this process by antibiotic therapy is believed to cause osmotic diarrhea. Some, but not all, bacteria can deconjugate bile salts, and unconjugated bile salts are known to stimulate fluid secretion by the colonic mucosa; another mechanism for AAD may be reduced bacterial degradation of bile salts within the colonic lumen. Other mechanisms that can account for AAD include stimulation of intestinal motility through the motilin-like effect of erythromycin, an allergic reaction, or infection with microorganisms other than Clostridium difficile, including Clostridium perfringens type A, Staphylococcus aureus, and Salmonella enterica.2–4
The genotype of C. perfringens that causes AAD appears to be distinct from those that induce food poisoning.3,5 Type A strains isolated from patients with AAD carry the C. perfringens enterotoxin (CPE) gene on a plasmid, whereas those that cause food poisoning have a chromosomal CPE gene. S. aureus was identified as a cause of severe AAD and enterocolitis before C. difficile-associated diarrhea was identified.2,6 Since the advent of sensitive and specific testing for C. difficile, however, very few cases of S. aureus AAD have been confirmed, and the true role played by this pathogen in AAD is unclear. Antibiotic-associated infection with Klebsiella oxytoca has been described in patients with right-sided hemorrhagic colitis. This rare pathogen releases several potent toxins, and it appears to colonize the bowel after the indigenous flora has been altered by exposure to antibiotics.7
AAD complicates 2% to 5% of antibiotic treatment courses, but the incidence varies depending on the antibiotic used; it is more common, for example, during therapy with ampicillin (5%-10%), amoxicillin-clavulanate (10%-25%), or cefixime (15%-20%) and less common during therapy with fluoroquinolones (1%-2%) or trimethoprim-sulfamethoxazole (<1%).8–9
Most cases of AAD are mild and self-limited. Pseudomembranous colitis is absent, and significant complications are rare. C. difficile infection accounts for less than 10% of AAD cases but is an important pathogen to identify because it often requires specific antimicrobial therapy and can lead to life-threatening complications, as discussed in the following section. A comparison between the clinical features of AAD caused by C. difficile and AAD from other causes is presented in Table 108-1.
Table 108-1 Differences between Antibiotic-Associated Diarrhea from Clostridium difficile Infection and from Other Causes
| CHARACTERISTIC | AAD FROM C. DIFFICILE INFECTION | AAD FROM OTHER CAUSES |
|---|---|---|
| Most commonly implicated antibiotics | Clindamycin, cephalosporins, penicillins, fluoroquinolones | Clindamycin, cephalosporins, ampicillin, or amoxicillin-clavulanate |
| History | Usually no history of antibiotic intolerance | History of diarrhea with antibiotic therapy is common |
| Clinical Features | ||
| Diarrhea | May be florid; evidence of colitis with cramps, fever, and fecal leukocytes is common | Usually moderate in severity (nuisance diarrhea) without evidence of colitis |
| Findings on CT or colonoscopy | Evidence of colitis is common; pseudomembranes often are present | Usually normal |
| Complications | Hypoalbuminemia, anasarca, toxic megacolon; relapse can occur after treatment with metronidazole or vancomycin | Usually none except occasional cases of volume depletion |
| Results of assay for C. difficile toxin | Positive | Negative |
| Epidemiologic pattern | May be epidemic or endemic in hospitals or long-term care facilities | Sporadic |
| Treatment | ||
| Withdrawal of implicated antibiotic | Condition can resolve but often persists or progresses | Condition usually resolves |
| Antiperistaltic agents | Contraindicated | Often useful |
| Oral metronidazole or vancomycin | Prompt response | Not indicated |
AAD, antibiotic-associated diarrhea; CT, computed tomography.
From Bartlett JG. Clinical practice: Antibiotic-associated diarrhea. N Engl J Med 2002; 346:334.
TREATMENT
Because AAD is believed to result from an alteration of the normal colonic microflora, a variety of probiotic agents has been evaluated for its treatment and prevention. In a double-blind controlled clinical trial, oral capsules containing viable Saccharomyces boulardii, a nonpathogenic yeast, were coadministered with antibiotics; this combination treatment reduced the incidence of AAD in hospitalized patients from 22% in the placebo group to 9.5% in the S. boulardii group (P = 0.04).10 Another randomized, placebo-controlled trial, however, failed to demonstrate a beneficial effect for S. boulardii in an elderly population of antibiotic recipients.11 Lactobacillus species, in particular Lactobacillus rhamnosus GG, also have been studied in clinical trials of AAD. In one study, Lactobacillus GG was effective in reducing the incidence of AAD to 5% in children being treated for respiratory tract infections compared with a 16% incidence in the placebo group12; other clinical trials of Lactobacillus GG have yielded negative results.13
A meta-analysis examined the results of randomized, double-blind, placebo-controlled trials of probiotic therapy for AAD published between 1966 and 2000.14 Nine studies were analyzed, including four using S. boulardii and four using Lactobacillus GG. The combined odds ratio for AAD in the probiotic-treated groups was 0.37 compared with placebo (95% confidence interval [CI]: 0.26-0.53; P < 0.001). For S. boulardii, the odds ratio in favor of active treatment over placebo was 0.39 (95% CI: 0.25-0.62, P < 0.001) and for lactobacilli the odds ratio was 0.34 (95% CI: 0.19-0.61, P < 0.01). A second meta-analysis yielded similar results.15 Thus, the weight of published evidence suggests that probiotic agents such as S. boulardii and lactobacilli, when used in combination with antibiotics, reduce the risk for AAD. Such therapy may be especially advantageous in patients with a history of susceptibility to AAD.
PSEUDOMEMBRANOUS ENTEROCOLITIS
A case report by Finney published in 1893 is considered to be the first description in the medical literature of pseudomembranous enterocolitis.9,16 In that instance, fatal pseudomembranous inflammation of the small intestine followed surgery in a debilitated young woman with gastric outlet obstruction caused by peptic ulcer disease. The presence of an inflammatory pseudomembrane overlying the intestinal mucosa characterizes pseudomembranous colitis (when the colon alone is involved) or pseudomembranous enterocolitis (when the small intestine also is involved).9 The pseudomembrane comprises inflammatory and cellular debris and forms distinctive patches of yellow or gray exudate that obscure the underlying mucosa. In early lesions, a 1- to 2-mm area of punctate ulceration may be visible. Classic pseudomembranes consist of ovoid plaques of 2 to 10 mm in diameter separated by areas of normal or hyperemic mucosa. Histologically, pseudomembranes can be seen to emanate from central areas of epithelial ulceration to form the mucosal plaques. In more-severe cases, the areas of ulceration and the overlying pseudomembranes can coalesce to cover large areas of mucosa.
Risk factors for the development of pseudomembranous enterocolitis in the absence of C. difficile infection include intestinal surgery, intestinal ischemia, and other enteric infections. During the 1940s to the 1970s, most reported cases of pseudomembranous enterocolitis occurred following abdominal or pelvic surgery.17,18 Bartlett has identified numerous descriptions of pseudomembranous enterocolitis in the medical literature associated with a wide variety of other intestinal disorders including Shigella infection, Crohn’s disease, neonatal necrotizing enterocolitis, intestinal obstruction, Hirschsprung’s disease, and colonic carcinoma.9 Intestinal ischemia can result in histologic changes similar to those observed in severe C. difficile colitis, although well-defined characteristic patchy pseudomembranes usually are not seen. Severe systemic insults including shock, advanced renal failure, spinal fracture, extensive burns, heavy metal poisoning, and hemolytic-uremic syndrome also have been associated with pseudomembranous enterocolitis. A potential common etiologic factor shared by many of these disorders is hypoperfusion of the intestinal mucosa leading to ischemic necrosis and ulceration.
Other infectious agents have been implicated as causes of pseudomembranous colitis in the absence of C. difficile infection, most notably S. aureus.2,3,6 Before C. difficile was identified as the most common cause of pseudomembranous colitis, S. aureus often was identified in stool cultures of patients with postoperative pseudomembranous enterocolitis, and oral vancomycin proved to be an effective therapy.6 In retrospect, it is difficult to ascertain whether the efficacy of vancomycin reflected its activity against staphylococcal infection or against unrecognized infection with C. difficile. Currently, 2% to 3% of patients with antibiotic-associated pseudomembranous colitis have negative tests for C. difficile and its toxins in stool specimens despite use of the most sensitive available assays; it remains unclear what proportion of these patients have false-negative tests for C. difficile or instead are infected with an as-yet-unidentified infectious agent.
CLOSTRIDIUM DIFFICILE-ASSOCIATED DIARRHEA AND COLITIS
C. difficile, an anaerobic, Gram-positive, spore-forming, toxigenic bacillus, was first isolated in 1935 from the fecal flora of healthy neonates.19 The organism passed into obscurity until 1978, when the association between toxins released by this organism and antibiotic-induced pseudomembranous colitis first was reported.20,21 Since that time, the incidence of C. difficile infection has increased dramatically, and the organism is now recognized as the primary cause of nosocomial infectious diarrhea in developed countries.22–27
The reported incidence of C. difficile-associated diarrhea has risen steadily over the past decade. For example, in the United States, the rate of nosocomial C. difficile infection per 100,000 population rose from 31 in 1996 to 61 in 200028 to 84 in 2005 (personal communication, L. Clifford McDonald, Centers for Disease Control and Prevention, Atlanta, Ga.). Similarly, C. difficile as a primary or contributing cause of death in the United Kingdom rose from 1000 cases in 2000 to 6500 cases in 2006.29 The reported incidence of community-acquired C. difficile AAD is substantially lower, ranging from 8 to 12 cases per 100,000 person-years.30,31
C. difficile infection also appears to be accompanied by greater morbidity and mortality in the past decade, owing in part to the emergence of increasingly virulent strains. One such strain, designated NAP1/BI, has a mutation in a bacterial gene called txcD, which allows the organism to produce more toxins. The mutant strain also produces a third toxin (called binary toxin) and is resistant to fluoroquinolones, making it more prevalent in patients receiving this class of antibiotics.32
PATHOGENESIS AND EPIDEMIOLOGY
The pathogenesis of C. difficile infection requires the following conditions: alteration of the normal colonic microflora by antibiotics or, rarely, chemotherapeutic agents; oral ingestion of C. difficile or its spores with resultant colonization of the large intestine; release of toxins A and B into the colonic lumen; binding and internalization of toxins by colonocytes; and subsequent colonic damage (colitis). Several host factors, particularly the immune response to C. difficile toxins, determine whether a patient remains an asymptomatic carrier or develops colitis (Fig. 108-1).33
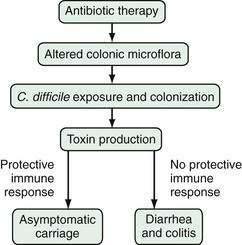
Figure 108-1. Pathogenesis of Clostridium difficile-associated diarrhea and colitis.
(From Kyne L, Farrell R, Kelly CP. Clostridium difficile. Gastroenterol Clin North Am 2001; 30:753.)
Alteration of the Colonic Microflora
Alteration of the resident colonic microflora, as a consequence of antimicrobial therapy, occurs in nearly all patients who develop C. difficile infection. The protective barrier provided by the intestinal microflora often is referred to as colonization resistance; its impairment by antibiotics and subsequent infection with C. difficile can be demonstrated in animal models.34,35 C. difficile also can colonize the intestines of germ-free mice but is eliminated after these animals are inoculated with fecal flora from normal mice, clearly confirming the importance of the normal flora in preventing colonization.34 Colonization resistance can be demonstrated in vitro by growth inhibition of C. difficile by fecal extracts from healthy adults but not by sterilized extracts.36
Human neonates and infants have poor colonization resistance because they have not yet developed a stable complex colonic microflora.37,38 Colonization rates with C. difficile of 25% to 80% have been reported in healthy infants and children up to 24 months of age, who, despite large concentrations of toxins in the feces, rarely develop C. difficile-associated diarrhea.39 Absence of toxin receptor expression on the immature colonic epithelium has been suggested as a mechanism to explain the carrier state in infants and children.40
Almost all antimicrobial agents can predispose to C. difficile diarrhea and colitis, including vancomycin and metronidazole,41,42 but the precise risks associated with individual agents are difficult to establish.43–45 The frequency of association of specific antibiotics is related to their frequency of use, their route of administration, and their effect on the colonic microflora.43,45 Antibiotics commonly associated with C. difficile infection and diarrhea include clindamycin, cephalosporins, ampicillin, amoxicillin, and (more recently) the fluoroquinolones (Table 108-2).32,44,46–48 Cancer chemotherapy agents that possess antibacterial properties and bowel preparation regimens (e.g., before colonoscopy or colonic surgery) rarely can result in sufficient disturbance of the intestinal microflora to allow subsequent colonization with C. difficile.49 One report indicated that healthy adults without known exposure to antibiotics or other modifiers of the colonic microflora also occasionally develop C. diffile colitis.30
Table 108-2 Antimicrobial Agents That Predispose to Clostridium difficile-Associated Diarrhea and Colitis
Adapted from Kelly CP, LaMont JT. Treatment of Clostridium difficile diarrhea and colitis. In: Wolfe MM, editor. Gastrointestinal Pharmacotherapy. Philadelphia: WB Saunders; 1993. p 199; and Bouza E, Burillo A, Muñoz P. Antimicrobial therapy of Clostridium difficile–associated diarrhea. Med Clin North Am 2006; 90:1141-63.
Hospital Epidemiology of Clostridium difficile Infection
Chronic intestinal carriage rates of C. difficile in healthy adults are low (0% to 3% in American and European populations) and might represent intestinal transit without true colonization.27,50,51 It also is unclear whether carriage is a temporary or permanent state.42
In contrast, hospital inpatients treated with antibiotics have reported colonization rates of 10% to 21%.23,27,52–55 The hospital environment is a major source of C. difficile infection; not only infected stool, but also environmental surfaces, soiled bedding, bedpans, toilet seats, and the hands and stethoscopes of health care workers are potential sources of nosocomial C. difficile infection.23,54 In one study, C. difficile was acquired on average in 3.2 days by patients who shared a room with an infected roommate compared with 18.9 days by patients in single rooms or with roommates whose stool cultures were culture negative for C. difficile.52 In the same study, C. difficile was cultured from the hands of 59% of hospital workers caring for patients with positive C. difficile cultures. The organism also was frequently cultured from bedrails, toilets, floors, call buttons, and other surfaces in the rooms of infected patients.
Asymptomatic carriers rarely develop C. difficile-associated diarrhea, but they serve as an important reservoir of nosocomial infection.55,56 In one study, 29% of environmental cultures taken from the hospital rooms of symptom-free carriers were positive for C. difficile, compared with only 8% of cultures from rooms of patients who were culture-negative for C. difficile.52
In antibiotic-treated animals, the infective dose of toxigenic C. difficile may be as low as two organisms.21 If human susceptibility is similar, control of C. difficile infection in hospitals will continue to be a major challenge because up to 109 organisms per gram are excreted in liquid feces.57,58 Highly resistant spores of C. difficile can persist for many months in the hospital environment and can result in infection if ingested by a susceptible host.57
Although it is not possible to eradicate C. difficile and its spores from the hospital environment, certain control measures have been recommended to reduce the prevalence of C. difficile-associated diarrhea (Table 108-3).59,60 Infected inpatients should be bedded in private rooms whenever possible to reduce patient-to-patient spread. Strict enteric precautions and regular hand washing after patient contact should be observed, because C. difficile can be cultured from the hands of health care workers after as many as 60% of contacts with infected patients.52 The use of alcohol-based hand gels is not as effective as washing with soap and running water in removing C. difficile spores.60 A controlled trial of using vinyl disposable gloves during patient contact also reduced the transmission of infection.61 After discharge of infected patients, surface environmental disinfection is best performed using a cleaning agent (e.g., hypochlorite solution) containing at least 5000 ppm available chlorine.60
Table 108-3 Practice Guidelines for the Prevention of Clostridium difficile Diarrhea
From Fekety R. Guidelines for the diagnosis and management of Clostridium difficile-associated diarrhea and colitis. American College of Gastroenterology, Practice Parameters Committee. Am J Gastroenterol 1997; 92:739.
Hospital outbreaks of C. difficile-associated diarrhea are common and likely result from the close approximation of susceptible persons (elderly and infirm patients) who are taking antibiotics and who are then exposed to the pathogen either in the hospital environment or through person-to-person spread. Some recent reports suggest that hospital and community outbreaks are related to the emergence of mutated hypervirulent strains, which are highly toxigenic and resistant to numerous antibiotics including fluoroquinolines.32,48 Prophylactic therapy with metronidazole or vancomycin is not effective as a disease control measure,62 and C. difficile diarrhea is prevented best by avoiding the unnecessary use of broad-spectrum antibiotics, especially in hospitalized patients, and by careful attention to hand hygiene.
In the future, increasing individual and herd immunity to C. difficile by vaccination or by passive immunotherapy may become a viable approach to reducing the prevalence of this common nosocomial disease.59,63–65 Prophylactic measures such as the use of bacterial and yeast probiotics or toxin binders in high-risk hospital patients also warrant further investigation.66–69
Clostridium difficile Toxins
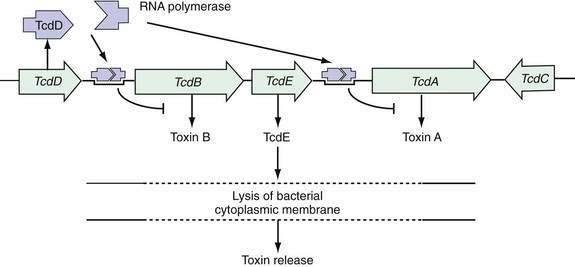
Pathogenic strains of C. difficile produce two structurally similar protein exotoxins, toxin A and toxin B, which are the only known virulence factors. The genes encoding toxin A and toxin B reside in a 19.6-kb chromosomal region, the C. difficile pathogenicity locus, which contains the genes encoding toxin A (tcdA) and B (tcdB) as well as two putative regulatory genes (tcdC and tcdD, also called tcdR) (Fig. 108-2).70–75 The tcdD gene product appears to up-regulate toxin transcription by complexing with RNA polymerase that binds to the toxin promoter regions. The tcdC gene is transcribed in the opposite direction to tcdA, tcdB, and tcdD, and its gene product appears to decrease toxin production.70–75 Mutations of tcdC are associated with increased virulence that may be related to increased toxin production.76,77 The fifth gene of the pathogenicity locus, tcdE, encodes a protein of undetermined function, although some data support the theory that it acts to lyse bacterial cell walls, thereby releasing toxins A and B.78
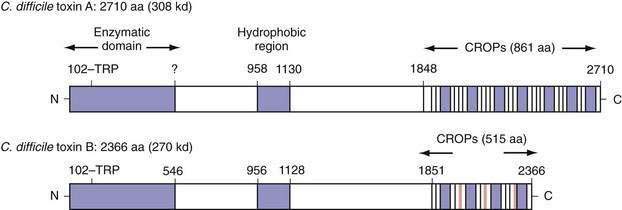
Toxins A (308 kd) and B (220 kd) are members of the large clostridial cytotoxin family, share a number of structural features, and are 49% identical at the amino acid level.79–81 Both toxins carry an N-terminal enzymatic domain that mediates their toxic effects on mammalian cells, a central hydrophobic region that might act as a transmembrane domain to facilitate entry into the cytoplasm, and a C-terminal domain consisting of a series of repeated sequences that mediate toxin binding (Fig. 108-3). Both toxins function as uridine diphosphate glucose (UDP-glucose) hydrolases and glucosyltransferases, a requirement for their cellular toxic effects.
Following internalization into the host cell cytoplasm, the toxins catalyze the transfer and covalent attachment of a glucose residue from UDP-glucose to a conserved threonine amino acid on small (20 to 25 kd) guanosine triphosphate–binding rho proteins. Rho proteins are part of the Ras superfamily, are expressed in all eukaryotic cells, and act as intracellular signaling molecules to regulate cytoskeletal organization and gene expression. The rho proteins, RhoA, Cdc42, and Rac, are substrates for both toxins A and B, whereas Rap is a substrate for toxin A only.74,82–84 Glucosylation of rho proteins by the toxins leads to disordered cell signaling, disorganization of the cytoskeleton, disruption of protein synthesis, cell rounding, and cell death.74,85 Both toxins also activate nuclear factor κB (NF-κB), mitogen-activated protein (MAP) kinases, and cyclooxygenase (COX)-2 in target cells, leading to the release of proinflammatory cytokines including interleukin (IL)-1β, tumor necrosis factor (TNF)-α, and IL-8.85,86 These cellular proinflammatory effects contribute to the marked intestinal inflammatory response evident in C. difficile-associated diarrhea and pseudomembranous colitis.
Toxin A initially was thought to be the only enterotoxin based on studies in animals85,87,88 whereas toxin B, an extremely potent cytotoxin, had minimal enterotoxic activity in animals. This suggested that toxin B did not contribute to diarrhea and colitis in humans,87,89–91 although studies on human colon show that, in fact, toxin B is 10 times more potent than toxin A in inducing in vitro colon injury.92,93 Furthermore, toxin A−/toxin B+ strains of C. difficile have been isolated from patients with diarrhea and pseudomembranous colitis,94–97 confirming that toxin B is a major virulence factor in human disease.
The Immune Response to Clostridium difficile
Serum IgG and IgA antibodies against C. difficile toxins are found in more than 50% of healthy children and adults.98–102 Mucosal IgA antitoxin antibodies also are detectable in more than 50% of human colonic secretions and might inhibit receptor binding of toxin A.100,102 Immunization against C. difficile toxins protects animals from C. difficile colitis but does not protect against colonization—a situation that may be similar to the asymptomatic carrier state in humans.53,103
High serum IgG antitoxin A antibody concentrations are associated with protection against C. difficile-associated diarrhea and colitis.63–65 Recurrent C. difficile diarrhea has been associated with low serum antitoxin antibody concentrations in children and in adults.98,102,104,105 In one study, adult inpatients with C. difficile diarrhea and a low level of serum IgG against toxin A had a 48-fold greater risk of recurrent disease after successful treatment compared with patients who had high antitoxin concentrations (Fig. 108-4).106 High serum IgG antitoxin A concentrations also have been identified in asymptomatic carriers of toxigenic C. difficile.
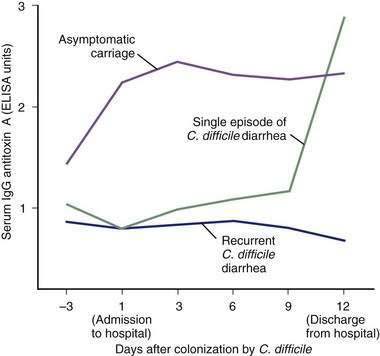
In a prospective study of nosocomially acquired C. difficile, the 51% of infected patients who were asymptomatic carriers had serum IgG antitoxin A concentrations that were three times higher than those in patients with diarrhea (see Fig. 108-4).53 The immune response to toxin B has not yet been clearly correlated with specific clinical outcomes. Nonetheless, toxin B is both pathogenic and immunogenic in humans, and antibody responses to toxin B might contribute to immune protection against C. difficile-associated diarrhea.63–65
Other Risk Factors for Clostridium difficile Infection
In addition to antimicrobial therapy, increasing age and increased comorbidity are important risk factors for C. difficile infection.107 In England and Wales, 75% of reported C. difficile infections between 1992 and 1996 occurred in patients aged 65 years or older.50 Data from the United States also demonstrate that age is an independent risk factor for this infection.108,109 The elderly particularly are predisposed to infection with C. difficile because of increased nosocomial exposure and frequent courses of antibiotics and a reduced ability of their polymorphonuclear leukocytes to phagocytose these organisms.110 In one study of antibiotic recipients, patients with severe underlying disease at the time of hospital admission were eight times more likely to develop C. difficile infection compared with patients who were less severely ill.53 Other reported risk factors for C. difficile infection include the use of a nasogastric tube, gastrointestinal procedures, intensive care unit stay, and length of hospital stay.43 The strengths of the associations of these risk factors with C. difficile vary from study to study. These factors often are markers of disease severity, older age, or both, and the significance of their association with C. difficile can decline or be lost after controlling for these confounding variables.53,107,109
The role of acid suppression in C. difficile infection is unclear. In theory, reduction of gastric acid could allow a greater number of viable spores to reach the colon; however, because spores are generally acid resistant, the importance of this effect is unclear. Some studies have shown an increased risk of C. difficile infection with acid suppression,109 but others have not confirmed this after adjusting for confounding variables.32,111
Patients undergoing cytotoxic chemotherapy for malignancy are at risk for C. difficile-associated diarrhea infection because of frequent antibiotic use, nosocomial exposure to C. difficile, and severe comorbidity.111,112 Even in the absence of antibiotic use, antineoplastic chemotherapy, especially with methotrexate, predisposes to C. difficile infection, reflecting the ability of these drugs to alter the colonic microflora and reduce C. difficile colonization resistance.49 C. difficile-associated diarrhea also has been reported in patients undergoing immunosuppressive therapy in the setting of solid organ or bone marrow transplantation.113,114
Patients with human immunodeficiency virus (HIV) infection also are at risk for C. difficile-associated diarrhea because of multiple risk factors, including frequent prophylactic and therapeutic antibiotic use, hospitalization, and immunocompromise.115–118 C. difficile colitis behaves the same in patients with acquired immunodeficiency syndrome (AIDS) as it does in control groups118 and testing for C. difficile should be a routine part of the diagnostic evaluation in patients with HIV infection, diarrhea, and a history of current or recent antibiotic treatment.
Patients with inflammatory bowel disease (IBD) are at increased risk for C. difficile infection.119–122 Infection with a broad range of enteric pathogens including C. difficile, Campylobacter, and Salmonella species can precipitate or mimic disease relapse in IBD. C. difficile is the most commonly identified specific pathogen in IBD patients in North America and Europe, however, and is present in as many as 5% to 19% of patients with relapse in some case series.119–122 Some IBD patients with C. difficile infection do not have a history of recent antibiotic use, suggesting that IBD itself might impair colonization resistance. The possibility of enteric infection with C. difficile or other pathogens should be considered in patients with an increase in IBD disease activity. If C. difficile infection is identified, antimicrobial therapy with metronidazole or vancomycin is indicated in combination with other IBD therapies. In one study, IBD inpatients with coexisting C. difficile infection were more likely to have severe disease and to require colectomy than similar patients without coexisting infection.122
CLINICAL FEATURES
Clinical manifestations of C. difficile infection range from asymptomatic carriage to mild or moderate diarrhea to life-threatening pseudomembranous colitis. Asymptomatic carriage of C. difficile is common in hospitalized patients. Several large epidemiologic studies indicate that 10% to 21% of hospital inpatients receiving antibiotics in high-risk units are carriers.28,52,53,55,123 Although most of the C. difficile isolates from carriers are toxin producing, the carriers do not develop symptomatic disease, perhaps as a result of protective immunity.28,55,104
In patients who develop diarrhea with C. difficile, symptoms usually begin soon after colonization. The incubation period is usually less than a week, with a median time of onset of approximately two days.28,52,53,124 Colonization can occur during or after antibiotic treatment. Olson and associates26 reported that 96% of patients with symptomatic C. difficile infection had received antibiotics within 14 days of the onset of diarrhea and that all had received an antibiotic within the previous three months.
C. difficile diarrhea typically is associated with the frequent passage of loose or watery bowel movements. Mucus or occult blood may be present, but melena or hematochezia is uncommon and, if present, should suggest the presence of IBD, colon cancer, or another source of bleeding. Some patients present with fever, leukocytosis, and cramping abdominal pain.125 Because C. difficile is not an invasive pathogen, extraintestinal manifestations of C. difficile infection such as septic arthritis, bacteremia, or splenic abscess are extremely rare.126–129 An oligoarticular, asymmetrical, nondeforming large-joint arthropathy, similar to that seen in other infectious colitides, sometimes is seen.130
Patients with more severe disease can develop a colonic ileus or toxic dilatation and present with minimal or even no diarrhea.125 In the absence of diarrhea, the only clues to the diagnosis may be high fever, moderate or marked (e.g., leukemoid) polymorphonuclear leukocytosis, lower or diffuse abdominal pain, tenderness, and distention.
Abdominal plain films might reveal a dilated colon (more than 7 cm in its greatest diameter), toxic megacolon, or small bowel ileus with air-fluid levels mimicking intestinal obstruction or ischemia. In such cases, a computed tomographic scan of the abdomen may reveal nonspecific features common to ischemic, infectious, and inflammatory colitides (Fig. 108-5).131 Radiologic features of pseudomembranous colitis include mucosal edema, a thickened colonic wall, pancolitis, and pericolonic inflammation with or without ascites, usually without any small bowel involvement other than ileus.132 Flexible sigmoidoscopy or colonoscopy is sometimes indicated to identify pseudomembranous colitis (see later) when the diagnosis remains unclear after initial evaluation.
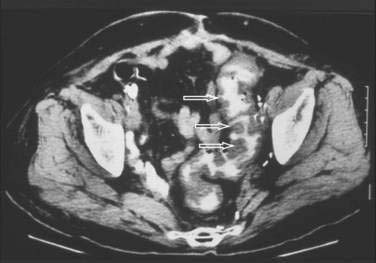
Complications of severe C. difficile colitis include dehydration, hypoalbuminemia, electrolyte disturbances, renal failure, hypotension, toxic megacolon, systemic inflammatory response syndrome, bowel perforation, and death.59,125
DIAGNOSIS
The diagnosis of C. difficile diarrhea or colitis is based on a history of recent or current antimicrobial therapy, development of diarrhea or other evidence of acute colitis, and demonstration of infection by toxigenic C. difficile, usually by detection of toxin A or B, or both, in a stool sample.48,59
Tests for Clostridium difficile Infection
The diagnosis of C. difficile diarrhea should be considered in any patient with acute diarrhea who has received antibiotics within the previous three months and especially in anyone whose diarrhea began 72 hours or more after hospitalization. Approximately 40% of patients with C. difficile diarrhea at tertiary referral centers are symptomatic on admission to the hospital, and most have had a recent prior hospitalization.52,53,133 Although a history of recent antibiotic use is common, it is not a requirement for diagnosis.30
Testing of solid or formed stools for C. difficile toxin is not recommended because only patients with diarrhea require treatment.52,53,59,62,123 Treatment of asymptomatic carriers with antimicrobial agents against C. difficile is not recommended because it might only prolong the carrier state beyond the usual two to six weeks.62 Follow-up stool testing is not indicated in an asymptomatic patient, even in patients discharged to chronic care facilities, because asymptomatic carriage is already highly prevalent in these facilities. Stool carriage of C. difficile can persist for up to six weeks after cessation of symptoms and does not require therapy.134 Because asymptomatic carriers can act as hidden reservoirs for C. difficile infection, especially in hospitals and nursing homes, universal precautions should be followed for all patients to reduce the likelihood of patient-to-patient spread of nosocomial infectious disease.
If C. difficile diarrhea is suspected, a freshly passed stool sample should be submitted immediately to the laboratory in a clean watertight container to be tested for the presence of fecal toxin A or B. Anaerobic storage or the use of transport media is not necessary, but storage at ambient temperatures can result in denaturation of fecal toxin; samples should therefore be tested immediately or refrigerated or frozen, pending later testing.54,135
A variety of laboratory tests are available to diagnose infection with toxigenic C. difficile
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


