Chapter 18 Alpha-1 antitrypsin deficiency and other metabolic liver diseases
1 Alpha-1 antitrypsin deficiency (α-1 ATD) is the most common metabolic liver disease in childhood. The diagnosis should be considered in all adults and children with chronic hepatitis or cirrhosis of unknown origin. α-1 ATD is associated with chronic liver disease in 10% of affected adults and in 10% to 15% of affected children.
2 Hereditary tyrosinemia is characterized by progressive liver failure, renal tubular dysfunction, and hypophosphatemic rickets. Patients are at high risk for hepatocellular carcinoma if the disease is untreated. Treatment is available if the disease is identified early in life.
3 Gaucher’s disease is the most common lysosomal storage disease. The clinical presentation and severity of liver involvement are variable.
4 Cystic fibrosis is the most common potentially fatal autosomal recessive disease in the white population. The prevalence of cirrhosis with portal hypertension is 10% to 20%.
Overview
2. In most cases, a diagnosis can be made with a complete history, physical examination, and appropriate laboratory studies; some diagnoses require a liver biopsy.
3. Genetic and metabolic liver diseases account for approximately 10% of liver transplans in children.
4. Liver transplantation (LT) should be considered in children with metabolic liver disease associated with failure to thrive, target organ dysfunction (e.g., central nervous system or kidneys) caused by a toxic metabolic product, or progressive liver failure.
5. The presence of one genetic mutation for a specific liver disease can modify the severity of other diseases. The heterozygous state of alpha-1 antitrypsin deficiency (α-1 ATD) may increase the risk of progression in hepatitis B virus (HBV) and hepatitis C virus (HCV) infections, nonalcoholic fatty liver disease (NAFLD), cystic fibrosis (CF), and cryptogenic cirrhosis. Genetic polymorphisms are potential modifiers of hepatic cirrhosis.
Alpha-1 Antitrypsin Deficiency (α-1 ATD)
Genetics
2. α-1 AT is encoded by a gene on the long arm of chromosome 14 (14q31-32.2); α-1 ATD is an autosomal co-dominant disorder affecting 1 in 1800 live births.
3. PiMM (Pi = protease inhibitor), the normal variant, is the phenotype present in 95% of the population and is associated with normal serum levels of α-1 AT.
4. More than 100 allelic variants of α-1 AT are recognized. Not all variants are associated with clinical disease.
5. The Z α-1 AT protein is caused by a single nucleotide substitution (Glu to Lys). The variant is most common in persons of northern European descent.
6. PiZZ and PiSZ phenotypes are associated with severe deficiency and liver disease, whereas the PiMZ phenotype leads to an intermediate deficiency.
Clinical Features
2. Liver involvement is often first identified in the newborn period as a result of persistent cholestatic jaundice. Affected infants tend to be small for gestational age. From 10% to 15% of persons with the PiZZ phenotype present with liver disease in the first years of life (Table 18.1).
 Of those presenting with neonatal liver disease, 10% to 30% develop moderate to severe liver disease with coagulopathy, poor growth, and ascites in childhood.
Of those presenting with neonatal liver disease, 10% to 30% develop moderate to severe liver disease with coagulopathy, poor growth, and ascites in childhood.
Of those presenting with neonatal liver disease, 10% to 30% develop moderate to severe liver disease with coagulopathy, poor growth, and ascites in childhood.
3. Serum aminotransferase, alkaline phosphatase, and gamma glutamyl transpeptidase (GGTP) levels may all be elevated.
4. Emphysema develops in 60% to 70% of adults with α-1 ATD over the age of 25 years, with the peak in the fourth and fifth decades.
TABLE 18.1 Findings in patients with alpha-1 antitrypsin deficiency (PiZZ or PiSZ phenotype)
| Infancy (1–4 mo) (%) | At 18 yr of age (%) | |
|---|---|---|
| Elevated serum alanine aminotransferase levels | 48 | 10 |
| Elevated serum gamma glutamyltranspeptidase levels | 60 | 8 |
| Clinical signs of liver disease | 17 | 0 |
From Sveger T, Eriksson S. The liver in adolescents with alpha 1-antitrypsin deficiency. Hepatology 1995; 22:514–517.
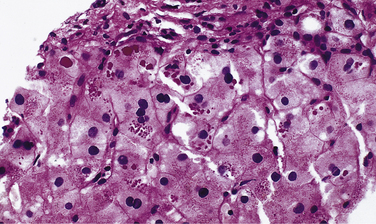
Pathogenesis and Diagnosis
1. Liver disease is associated with retention of abnormally folded Z protein in the endoplasmic reticulum of hepatocytes. Liver disease occurs in the PiZZ and PiSZ phenotypes but rarely in persons with PiMZ. Liver disease does not occur with the other variants (e.g., PiSS).
2. Far fewer patients exhibit liver and lung disease associated with α-1 ATD than estimated by population human genetic estimations, a finding suggesting involvement of unidentified genetic and environmental factors and modifier genes in the development of tissue damage.
3. The pathogenesis of α-1 ATD–associated liver disease is not completely understood. The following theories have been proposed:
 Accumulation of mutant protein in the endoplasmic reticulum may result in hepatotoxicity. This theory is supported by a transgenic mouse model and a study demonstrating delayed protein degradation of mutant α-1 AT Z protein in individuals with liver disease compared with those without liver disease.
Accumulation of mutant protein in the endoplasmic reticulum may result in hepatotoxicity. This theory is supported by a transgenic mouse model and a study demonstrating delayed protein degradation of mutant α-1 AT Z protein in individuals with liver disease compared with those without liver disease.
Accumulation of mutant protein in the endoplasmic reticulum may result in hepatotoxicity. This theory is supported by a transgenic mouse model and a study demonstrating delayed protein degradation of mutant α-1 AT Z protein in individuals with liver disease compared with those without liver disease.


4. The PiMZ state may predispose to more severe liver injury in various hepatic disorders (HBV and HCV infections, alcoholic liver disease, CF-associated liver disease, NAFLD).
5. The diagnosis is established by a serum α-1 AT level, phenotype (Pi typing), or genotype.
 Serum levels of serum α-1 AT are generally decreased in affected patients; however, α-1 AT is an acute phase reactant and can be falsely elevated. Serum concentrations are rarely higher than 50 to 60 mg/dL in patients with the PiZZ phenotype.
Serum levels of serum α-1 AT are generally decreased in affected patients; however, α-1 AT is an acute phase reactant and can be falsely elevated. Serum concentrations are rarely higher than 50 to 60 mg/dL in patients with the PiZZ phenotype.
Serum levels of serum α-1 AT are generally decreased in affected patients; however, α-1 AT is an acute phase reactant and can be falsely elevated. Serum concentrations are rarely higher than 50 to 60 mg/dL in patients with the PiZZ phenotype.
Treatment and Screening
2. Infants with cholestasis may benefit from fat-soluble vitamin supplements (vitamins A, D, E, and K) and infant formula containing medium-chain triglyceride oil. In addition, treatment with ursodeoxycholic acid may increase bile flow and reduce liver injury associated with cholestasis, although no evidence indicates a direct long-term benefit in α-1 ATD.
3. Avoidance of cigarette smoking, including second-hand smoke, and of environmental pollution exposure is mandatory to slow the progression of lung disease. Replacement therapy with purified or recombinant α-1 AT by inhalation or infusion was successful in slowing the decline in forced expiratory volume in a nonrandomized trial, and this therapy is often used.
4. LT is the recommended treatment for α-1 ATD–associated end-stage liver disease and liver failure. The recipient assumes the donor Pi phenotype and is no longer at risk for emphysema. Long-term survival is excellent. LT should be pursued before lung decompensation precludes transplantation.
5. Somatic gene therapy, in which a normal α-1 ATD gene is transferred to an organ capable of synthesizing the mature protein that could be secreted into the circulation, is potentially useful for the treatment of lung disease. Gene therapy for treatment of liver disease requires delivery of peptides to the endoplasmic reticulum to prevent polymerization of mutant protein or manipulation of the degradation system in those at risk for liver disease. The technology is currently limited by poor transfer of gene products and unknown safety risks.
6. Small molecule pharmacologic chaperone therapy and manipulation of autophagy are currently being evaluated as possible future treatment strategies.
Hereditary Tyrosinemia
Genetics
1. This disease is caused by a deficiency in the fumarylacetoacetate hydrolase (FAH) gene, the terminal enzyme in phenylalanine and tyrosine degradation.
2. This autosomal recessive defect has an incidence of 1 in 100,000. The disorder is most prevalent in French Canadians in Quebec, Canada, where it has an incidence of 1 in 1800.
Clinical Features
1. The disorder is characterized by progressive liver failure, renal tubular dysfunction, and hypophosphatemic rickets.
2. It may manifest as acute hepatic failure in infancy, neonatal cholestasis, rickets, or failure to thrive, or, later in childhood, as compensated or decompensated cirrhosis. The acute form usually manifests with poor growth, irritability, and vomiting. Death from liver failure by 1 to 2 years of age is not uncommon in untreated patients.
3. Patients have a characteristically prolonged prothrombin time despite mild elevations in aminotransferase and bilirubin levels. Serum alkaline phosphatase levels may be disproportionately elevated because of rickets caused by renal tubular involvement.
4. Neurologic crises develop and resemble acute intermittent porphyria, presumably from competitive inhibition of δ-aminolevulinic acid (ALA) dehydratase by succinylacetone.
5. Cardiomyopathy, particularly interventricular septal hypertrophy, is found in 30% of newly diagnosed patients. This complication resolves during treatment in most patients.
Pathogenesis and Diagnosis
1. Tyrosine metabolites, including tyrosine and succinylacetone, proximal to the FAH blockage accumulate.
2. Succinylacetone and succinylacetate inhibit enzymes, including porphobilinogen synthase, and this process results in increased levels of ALA, which is responsible for acute neurologic crises.
4. Liver histology is characterized by macrovesicular steatosis, pseudoacinar formation of hepatocytes, hemosiderosis, and variable hepatocyte necrosis and apoptosis. Periportal fibrosis progresses to micronodular cirrhosis with regenerative nodules.
5. The diagnosis is established by the presence of elevated levels of succinylacetone in the urine or by genotyping. Other features include elevated plasma levels of tyrosine, methionine, and alpha fetoprotein, which are nonspecific findings. The diagnosis should be considered in patients with cirrhosis who have diminished hepatic synthetic function with mildly elevated aminotransferase levels. Renal tubular dysfunction results in glycosuria, proteinuria, amino aciduria, and hyperphosphaturia.





